Revisión
Patología Estructural en Enfermedades Glomerulares Primarias
Structural pathology in primary glomerular diseases
Actual Med.2024;109(820):176-186 DOI: 10.15568/am.2024.820.rev02
Recibido: 19/01/2023
Revisado: 15/03/2023
Aceptado: 18/11/2024
RESUMEN
La biopsia renal percutánea se ha convertido en parte de la práctica clínica en el estudio de las nefropatías, así como ha revolucionado el manejo de estas enfermedades y es útil para determinar el pronóstico de las mismas. Las Indicaciones más comunes incluyen el síndrome nefrítico y nefrótico, para el diagnóstico de lesiones renales primarias y la evaluación de pequeñas masas renales. Los pacientes con glomerulopatías primarias se pueden presentar con un proceso agudo o crónico, con daño limitado al riñón o con alteraciones en otros órganos, algunos de los síndromes que se observan durante la enfermedad son el nefrótico, nefrítico y glomerulitis rápidamente progresiva. Se describen los patrones histológicos de cada glomerulopatía primaria como la Enfermedad de Cambios Mínimos, Glomeruloesclerosis focal y segmentaria, Glomerulopatía membranosa, Glomerulonefritis postinfecciosa, Glomerulopatía membrnaoproliferativa y Nefropatía por IgA. También se describen algunos nuevos biomarcadores histológicos, serológicos y de orina de cada patología para su diagnóstico, pronóstico y tratamiento específico. Conclusiones. El diagnóstico específico de las enfermedades glomerulares primarias debe incorporar información relevante de la historia clínica, así como estudios genéticos, histopatológicos, análisis serológicos y urinarios.
Palabras clave: Biopsia renal; Glomerulopatías primarias; Patología estructural; Biomarcadores histopatológicos.
ABSTRACT
Percutaneous renal biopsy has become part of clinical practice in the study of nephropathies, as well as revolutionizing the management of these diseases and is useful for determining their prognosis. The most common indication of renal biopsy includes nephritic and nephrotic syndrome, for the diagnosis of primary renal lesions and the evaluation of small renal masses. Patients with primary glomerulopathies can present with an acute or chronic process, with damage limited to the kidney or with alterations in other organs. Some of the syndromes observed are nephrotic, nephritic, and rapidly progressive glomerulonephritis. The histological patterns of each primary glomerulopathy are describe, such as minimal change disease, focal segmental glomerulosclerosis, membranous glomerulopathy, postinfectious glomerulonephritis, membranoproliferative glomerulopathy and IgA nephropathy. Some new histological, serological and urine biomarkers of each pathology for their diagnosis, prognosis and specific treatment are also described. Conclusions. The specific diagnosis of primary glomerular diseases must incorporate relevant information from the clinical history, as well as genetic, histopathological, serological and urinary tests.
Keywords: Renal biopsy; Primary glomerulopathies; Structural pathology; Histopathological biomarkers.
Leer Artículo Completo
INTRODUCCIÓN
En la patología estructural se utilizan técnicas moleculares, microbiológicas, inmunológicas y morfológicas para intentar explicar las causas de los signos y síntomas manifestados por los pacientes(1).
Con la descripción histológica de cada enfermedad podemos entender la clínica y también realizar el diagnóstico diferencial entre cada glomerulopatía. Continuamente se están realizando investigaciones para encontrar nuevos biomarcadores en suero, orina o tejido renal que apoyen el diagnóstico, se identifiquen como diana terapéutica o den a conocer el pronóstico de cada enfermedad.
Las glomerulopatías se pueden clasificar en primarias, secundarias, hereditarias y no clasificadas de acuerdo a patrón histológico y presentación clínica. En un estudio de biopsias renales de pacientes pediátricos con enfermedad glomerular se encontró que el 72% presentaba una enfermedad primaria, el 23% secundaria, 4% congénita y 0.5% no clasificada(2,3). La interpretación de la biopsia renal debe de tomar en cuenta el origen étnico, edad y presencia de hipertensión arterial al momento de toma de la muestra, ya que estos factores pueden modificar el valor de la histología renal(4,5). La complicación más común es hematoma perirenal autolimitado(6,7).
PATOLOGÍA ESTRUTURAL EN ENFERMEDADES GLOMERULARES PRIMARIAS
Síndromes clínicos en las enfermedades glomerulares
Síndrome Nefrótico
Se define por la presencia de proteinuria en rango nefrótico, hipoalbuminemia y edema(8).
La alteración renal se encuentra en la barrera de filtración glomerular en la abertura del diafragma proteico entre los podocitos o por una fosforilación disminuida de la nefrina. La pérdida de albúmina se ha atribuido a un defecto en la podocalixina que es una proteína cargada negativamente y puede reducir el diámetro de las fenestraciones y así impedir su filtración por el endotelio(9).
Las glomerulopatías que se asocian con más frecuencia al síndrome nefrótico son la Enfermedad por Cambios mínimos (ECM), Glomeruloesclerosis Focal y Segmentaria (GEFyS) y glomerulonefritis membranosa (GM)(8).
Síndrome Nefrítico
Se caracteriza por la presencia de hematuria, hipertensión, oliguria y edema. Es la presentación más común en las glomerulopatías proliferativas(10).
En el examen general de orina se observan eritrocitos dismórficos, acantocitos, cilindros hemáticos (en ocasiones de leucocitos) y proteinuria (no nefrótica). El número de cilindros hemáticos observados tiene una fuerte correlación con el número de semilunas observadas en la biopsia(11,12).
Se cuenta con una disrupción en la estructura de la barrera de filtración glomerular causada por varios mecanismos: daño directo de células endoteliales, depósito de inmunocomplejos, disrupción en MBG o daño en los podocitos(10).
Glomerulonefritis rápidamente progresiva
Es un síndrome clínico que se manifiesta por signos de daño glomerular (nefritis) a nivel urinario, junto con un deterioro progresivo y rápido de la función renal, en un periodo que puede comprender desde días, semanas o meses(13,14).
Presenta hematuria de manera abrupta, proteinuria, anemia y falla renal progresiva. Se puede presentar con fatiga, fiebre, pérdida de apetito, pérdida de peso. Hematuria ocasionalmente macroscópica, la microcópica es acompañada de eritrocitos dismórficos, cilindros hemáticos(14).
Histológicamente se caracteriza por el desarrollo de semilunas epiteliales, necrotizantes glomerulares. Fisiopatológicamente, el daño se origina en las paredes de los capilares glomerulares, lo cual induce una respuesta inflamatoria a nivel del espacio de Bowman y finalmente conducirá al desarrollo de semilunas fibroepiteliales y fibrosas(13). El diagnóstico histopatológico es con la presencia de glomerulonefritis proliferativa necrotizante. Se divide en tres tipos de acuerdo a IF: tipo 1: linear, tipo 2: granular y tipo 3: pauci-inmune(14). Linear refiere una enfermedad anti-MBG, granular a enfermedades con inmunocomplejos circulantes y pauci-inmune es referente a vasculitis asociadas a ANCA(14).
Se clasifican por mecanismos etiopatogénicos: Tipo 1: Por anticuerpos anti-MBG. Tipo 2: Por depósito de inmunocomplejos. Tipo 3: pauci-inmune. IF y ME negativa a depósitos inmunes. Tipo 4: anticuerpos doblemente positivos. Idiopática: Tipo 2 que no cumple todos los criterios y tipo 3 ANCA-negativo(13).
Patrones histopatológicos en Enfermedades Glomerulares
Enfermedad por Cambios Mínimos y Glomeruloesclerosis Focal y Segmentaria
La ECM es la mayor causa de síndrome nefrótico representando el 70% al 90% de los pacientes(15).
La patogénesis se da por un desequilibrio entre CD4+, CD8+, y células NK, así como una producción local de oxidantes tóxicos para los podocitos que al ser estimulados expresan el TLR-4 y CD80 para iniciar una respuesta inmune, así se puede iniciar una redistribución y pérdida de la nefrina y aumenta la permeabilidad a la albúmina(16).
En el microscopio óptico (MO) se observa una estructura normal.
La inmunofluorescencia (IF) es usualmente negativa, puede observarse tinción para IgM en el área mesangial de baja intensidad acompañado de C3 o C1q o encontrarse depósitos glomerulares de IgA (nefropatía por IgA con ECM). Una distribución segmentaria de tinción para IgM y C3 sugiere fuertemente una GEFyS aún sin presentar lesiones escleróticas(17).
En la microscopía electrónica (ME) se observa un extenso borramiento de los pedicelos, con vacuolización y transformación microvellosa de las células epiteliales(15).
En la GEFyS durante las faces iniciales hay activación y migración de las células epiteliales parietales (PECs) desde la cápsula de Bowman al penacho glomerular y esto puede inducir a la esclerosis del glomérulo. Los primeros glomérulos afectados son los yuxtamedulares(17,18).
En la MO se observa un patrón de cicatrización donde la esclerosis afecta a algunos glomérulos (focal) y a una porción del penacho glomerular (segmentaria). Una hipertrofia anormal de los glomérulos es un indicador precoz del proceso esclerótico. El proceso esclerótico se define por el colapso de los capilares glomerulares con aumento de la matriz y varía desde lesiones precoces pequeñas hasta una esclerosis casi global, las lesiones segmentarias son definidas y pueden localizarse a nivel perihiliar y/o en porciones periféricas del glomérulo. La glomeruloesclerosis se asocia a atrofia tubular y a fibrosis intersticial con linfocitos intersticiales(17).
La IF muestra un atrapamiento inespecífico de IgM y C3 en áreas de esclerosis o en zonas de aumento de la matriz mesangial(17).
La ME muestra borramiento en los procesos de los pedicelos de una manera difusa(6). La medición del ancho de los procesos es significativamente mayor en pacientes con GEFyS primaria a comparación con los pacientes con GEFyS secundaria y de etiología genética, así mismo el borramiento de estos procesos pedicelares es difuso y más severo en GEFyS primaria y se observa el borramiento de forma segmentaria en GEFyS secundaria y de causa genética(19).
La GEFyS es una lesión histológica renal con diferentes causas y patogenia, tiene varias subclases: primaria (por factores circulantes), genética y secundaria (maladaptación, virus o medicamentos)(19).
La GEFyS se puede presentar histológicamente en el MO con diferentes variantes:
Llesión en la punta: Afecta exclusivamente al polo tubular del glomérulo con una lesión que afecta al 25% de la región con adherencias entre el penacho y la cápsula de Bowman(17).
Celular: Se observa proliferación endocapilar que afecta al menos al 25% del penacho y ocluye la luz, se considera una lesión activa precoz de GEFyS(17).
Perihiliar: Esclerosis y hialinosis perihiliares que afectan a >50% de los glomérulos afectados. Son frecuentes las glomerulomegalias y las adherencias, así como habitual encontrar hialinosis arteriolar(17).
Se cree que la GEFyS primaria se debe a un factor o factores circulantes, algunos se han propuesto como candidatos específicos, ninguno aún válido, que alteran la permeabilidad podocitaria: el Receptor soluble del activador de plasminógeno de tipo uroquinasa (suPAR) puede ser pronóstico para la enfermedad renal crónica. La Citocina-1 similar a la cardiotrofina tiene la capacidad para disminuir la expresión de nefrina. La Angiopoyetina-like-4, se ha observado en niveles más altos en suero y podocitos con síndrome nefrótico y proteinuria masiva, pero no hace distinción entre ECM, GEFyS y GM(20).
Se observó una expresión anormal de miR-191 y miR-151-3p en tejido renal de niños con síndrome nefrótico observándose el primero aumentado significativamente de 3 a 5 veces más que los controles sanos y encontrando miR-151-3p disminuido significativamente en los pacientes con síndrome nefrótico. Se encontró también que la expresión de miR-150 fue significativamente menor en ECM(21).
Los podocitos expresan CD80 (también conocido como B7.1) en circunstancias específicas que pueden estar vinculadas con la proteinuria, la reorganización de la actina y la migración de podocitos. Se ha encontrado que los niveles en orina de CD80 son constantemente altos en pacientes con síndrome nefrótico cortico-sensibles y en episodios de recaída en ECM comparados con pacientes en remisión o con GEFyS, los niveles elevados de CD80 están asociados a un buen pronóstico a largo plazo(22).
CD44 se ha considerado un marcador de PECs activadas, al encontrarse positivo se considera de mal pronóstico o recuerrencia en pacientes pediátricos con GEFyS, lo cual puede ayudar a diferenciar GEFyS de ECM(18).
Nefropatía por IgA
Se da por un sistema inmune defectuoso al presentar una IgA1 pobre en O-galactosidasa producida en mucosas; se libera por respuesta del sistema inmune innato a través de la unión a receptores tipo Toll (TLR), la activación de células B y la sobreexpresión de TLR9 en las amígdalas como resultado de infecciones en mucosas(4,23). Los depósitos de inmunocomplejos mesangiales pueden llegar vía directa por activación de complemento o por una interacción con receptores específicos mesangiales, pueden ser de IgA1 galactosidasa-deficiente, IgA1/IgG o IgA/IgA, inducen la activación, proliferación, mayor síntesis de matriz mesangial y eventualmente lesión celular(24). Los depósitos de IgA mesangiales pueden activar el complemento por dos vías, una con presencia de C3 (vía alternativa) con depósitos de lectina unidora de manosas mesangial y por vía de las lectinas con depósitos de C4d que se asocia con mayor daño renal(24).
Al MO se observa desde una expansión mesangial mínima hasta una lesión proliferativa difusa con semilunas o esclerosis difusa. Hay aumento de la superficie mesangial por incremento de células de la matriz y por los depósitos que son una interposición mesangial y escisión de la MBG. En los casos crónicos se observa una esclerosis segmentaria con atrofia tubular y fibrosis intersticial proporcionales(17).
La IF muestra depósitos, de manera difusa y global de IgA en el mesangio o que se extienden a las asas capilares periféricas asociadas a las lesiones proliferativas. El C3 casi invariable esta presente, C1q puede o no estar presente. Puede haber depósitos de lambda predominantemente o IgG y/o IgM(17).
Al ME los depósitos se localizan en las zonas del mesangio por debajo de la MBG. En ocasiones hay depósitos subepiteliales e intramembranosos. Los pedicelos están borrados en las regiones de esclerosis. La distribución de los depósitos equivale al patrón de IF(17).
La medición del biomarcador Gd-IgA1 (IgA1 deficiente en O-galactidasa) se puede realizar en suero, orina o con la presencia en células glomerulares. Es un biomarcador específico de la enfermedad, sin embargo, aún no se cuentan con valores establecidos(23). Los niveles séricos de anticuerpos específicos IgG contra Gd-IgA1 están significativamente asociados con la progresión y recurrencia de la enfermedad(23). Otro biomarcador es la presencia de depósitos mesangiales de C4d, que se observa de manera temprana y está asociado con aumento y persistencia de la actividad, así como un peor pronóstico en la función renal(24).
Glomerulonefritis membranosa (GM)
Afecta a los podocitos con anticuerpos contra antígenos instrínsecos podocitarios (Receptor de fosfolipasa A2 de tipo M (PLA2R), el dominio trombospondina-like 7A (THSD7A), NEP y LRP2). Los anticuerpos anti-PLA2R y THSD7A están presentes en 50-80% y 2-4% de pacientes respectivamente. La clínica puede ser muy variable y se puede clasificar en primaria, con presencia (o no) de anticuerpos anti-PLA2R o anti-THSD7A o asociada a otras enfermedades o exposición a drogas o toxinas(4).
Los depósitos se localizan en la superficie podocitaria y posteriormente se liberan al espacio extracelular donde se adhieren a los componentes de la MBG(25).
En la MO se pueden encontrar pequeñas áreas lúcidas con la plata de Jones, que indican la falta de positividad a la plata a nivel del depósito, estos “agujeros” constituyen la primera manifestación de la glomerulopatía. Al progresar la reacción de la MBG, la matríz rodea los depósitos y determina una escisión a modo de enrejado o aspecto en escalera con técnicas de plata. Pueden aparecer lesiones adicionales que van desde semilunas hasta esclerosis. Lo que se asocia a un peor pronóstico es la presencia de esclerosis segmentaria, fibrosis intersticial y la atrofia tubular(17).
Con IF los depósitos característicos subepiteliales se visualizan como una positividad granular global y difusa a lo largo de la pared capilar, por lo general predomina IgG, suele estar presente C3. IgG4 es la subclase que se encuentra predominante en la GM idiopática, mientras IgG1, IgG2 e IgG3 se encuentran en casos de GM secundaria, si se reconocen IgA, IgM y C1q se debe plantear secundaria a Lupus Eritematoso Sistémico (LES)(17,25).
La ME muestra los depósitos electro-densos subepiteliales que al acumularse pueden observarse como poros delgados. La MBG se observa con la progresión de la membrana engrosada con los depósitos que se incorporan gradualmente a una nueva membrana y se pueden observar más electro-lúcidos hasta que se reabsorben completamente y desaparecen(17).
Existe un tipo de GM secundaria descrita en menores de 5 años causada por un antígeno alimentario, la albúmina sérica bobina catiónica (BSA), estos pacientes tienen altos niveles de anticuerpos anti-BSA (TgG1 e IgG4) que reaccionan a un péptido de BSA pero no al péptido homólogo en la sero-albúmina humana, el PLA2R sérico es negativo; cuentan con aumento de la permeabilidad en la barrera intestinal presentando episodios frecuentes de gastroenteritis(25).
Se han reportado casos sobre una nueva glomerulopatía relacionada con apolipoproteína E (ApoE) conocida como Enfermedad por depósitos de ApoE tipo GM. Estos casos muestran en la biopsia formaciones tipo espículas en la tinción de plata y depósitos electrodensos subepiteliales y subendoteliales, los depósitos se aprecian como microburbujas o microquistes. La IF no es específica y se ha identificado por inmunohistoquímica y espectrometría en masa la acumulación glomerular de ApoE(26).
Glomerulonefritis membranoproliferativa (GNMP)
La GNMP puede presentarse como primaria (principalmente en niños) o secundaria.
Se clasifica recientemente por el tipo de inmunocomplejos encontrados en la inmunofluorescencia(27, 28). En la fisiopatología puede ser mediada por inmunocomplejos o mediada por complemento por una alteración en la vía alterna.
En MO se observa expansión difusa del mesangio con hipercelularidad mesangial, por proliferación endocapilar e incremento de la matriz mesangial y engrosamiento y remodelamiento de las paredes capilares, con formación de doble contorno y duplicación de la MBG a menudo con un aspecto escindido en “vías de tren” con depósitos de inmunocomplejos(17, 28).
La GNMP se ha dividido en tres subtipos. El tipo I (mesangiocapilar) se produce una proliferación endocapilar difusa global con aumento de la celularidad y la matriz mesangial y simplificación del lóbulo. La proliferación suele ser uniforme y difusa, los glomérulos aparecen mas sólidos y nodulares. Con las técnicas de plata la pared capilar está engrosada con doble contorno. Este aspecto se debe a la presencia de depósitos subendoteliales y a la interposición del mesangio, según la cual las células mesangiales, los mononucleares del infiltrado e incluso parte de las células endoteliales se interponen entre el endotelio y la MBG con depósito de nuevo material de membrana basal interna. Este proceso genera un aspecto de doble contorno de la membrana basal circunferencial o parcial(17).
En la IF existen IgG, IgM y C3 de forma irregular y grumosa a nivel capilar y mesangial. La IgA es rara de encontrar. En la GNMP secundaria puede predominar la tinción con C3. Los depósitos del asa periférica adoptan una forma típica de salchicha y tienen un margen externo liso porque son subendoteliales y se amoldan por debajo de la MBG(17).
Al ME, la GNMP de tipo I muestra numerosos depósitos subendoteliales y mesangiales. Los depósitos se encuentran dentro de MBG inmediatamente por debajo de la lámina densa original(17).
La GNMP por complejos inmunes es causada por depósito creados por paraproteinemias (actualmente conocido como la causa principal de GNMP), enfermedades autoinmunes o infecciones crónicas(28).
Glomerulopatía C3 y Enfermedad por depósitos densos (DDD)
La glomerulopatía por C3, tiene mal pronóstico, es causada por activación, depósito y/o degradación anómala del complemento en la vía alterna. Se caracteriza por niveles séricos bajos de C3 persistentes. La IF muestra un predominio de tinción para C3(28).
En la ME la glomerulopatía por C3 muestra los depósitos en el mesangio, subendotelio, subepitelio y/o intramembranoso; en la DDD, se encuentran en el mesangio e intramembranosos altamente electrodensos. La principal entidad que representa la glomerulopatía por C3 es la DDD(28).
Glomerulonefritis postinfecciosa (GNP)
La GNP es una lesión glomerular inmunológica que ocurre como resultado a la respuesta de un huésped a una infección extrarenal. La glomerulonefritis más común es la postestreptocóccica; otras ocurren durante procesos bacterianos activos como endocarditis, abscesos profundos y comunicaciones arterioventriculares infectados en hidrocefalia. Inicia comúnmente con un síndrome nefrítico, o como proteinuria o hematuria aisladas(17,29).
Los inmunocomplejos que se depositan en el glomérulo pueden ser complejos de antígeno-anticuerpo del estreptococo o complejos autólogos. No hay correlación entre la cantidad de inmunocomplejos circulantes y la severidad de la glomerulopatía(29). La presencia de estreptococos activa directamente la vía alterna de complemento e inicia la formación de factor B, probablemente el factor B y las proteínas de estreptococos comparten algún epítope conformacional en común, lo que promueve la emergencia de autoanticuerpos anti-Factor B transitorios contribuyendo a la sobreactivación de complemento y depósito de inmunocomplejos con C3 en el glomérulo, éstos se depositan dentro o alrededor de la MBG reclutando células inflamatorias, también se activa la cascada de coagulación creando microtrombos(10,30). La presencia de anticuerpos anti-factor B hacen el diagnóstico diferencial con la glomerulopatía por C3 aunque estos autoanticuerpos desaparecen junto con la elevación de C3 en la evolución de la enfermedad(30).
Se han identificado dos proteínas, el “receptor de plasmina asociado a nefritis” (NAPlr) y “exotoxina B pirogénica estreptocóccica” (SPEB). NAPlr se ha identificado en los grupos A y C estreptocóccicos, los depósitos glomerulares ocurren al inicio de la enfermedad y son detectados en las biopsias localizados en las células mesangiales, endoteliales y neutrófilos. NAPlr es un gliceraldeido-3-fosfato deshidrogenasa con capacidad de unión a plasminógeno, una propiedad nefitogénica que ayuda al depósito de los inmunocomplejos circulantes. SPEB es un super antígeno producido por los estreptococos del grupo A con propiedades de unión a plasminógeno, es secretada como una exotoxina y se localiza en el glomérulo junto con C3 en las jorobas subepiteliales. Ambos antígenos activan la vía alterna del complemento resultando en niveles séricos bajos de complemento, promueven la formación de inmjnocomplejos y promueven el depósito de estos en el subendotelio(31).
En el MO usualmente se observan todos los glomérulos afectados de una extensión similar (difuso y global). Los capilares glomerulares se encuentran dilatados y con hipercelularidad sin evidencia de necrosis. Se puede observar un gran número de granulocitos. En algunas ocasiones con la tinción de tricómico de azul de metileno se pueden observar pequeños nódulos a una gran escala con inmersión en aceite, típicamente llamados “jorobas”(17,29).
La IF puede presentar depósitos granulares irregulares y difusos de IgG y C3 en las paredes capilares glomerulares, se describe como “cielo estrellado”. En la mitad de los casos se encuentra IgM, pero IgA, IgE y C1q normalmente están ausentes(17,29).
La MO muestra hinchazón del endotelio glomerular y células mesangiales. La MBG puede observarse engrosada y puede contener áreas electro-lúcidas posiblemente representado los depósitos ya resueltos. El signo clásico es la presencia de depósitos electro-densos subepiteliales de tipo cono que se llaman “jorobas”, localizadas cerca del mesangio, se encuentran de 4 a 5 jorobas por capilar y son más largas que las que se encuentran en la GM(17,29). La presencia de las jorobas tiene una correlación positiva entre la hipercelularidad glomerular y la formación de las jorobas subepiteliales. Los depósitos subendoteliales aparecen al inicio de la enfermedad y generalmente desaparecen a las 6 semanas. La fase de recuperación de la glomerulonefritis postinfecciosa muestra depósitos aislados mesangiales y ocasionalmente jorobas subepiteliales con áreas electro-lúcidas(29).
Existe una variante de GNP que se presenta en la IF positiva para IgA en lugar de C3. La histología típica para GNP está presente. El organismo más común que se ha aislado en estos pacientes es el estafilococo(31).
Vasculitis asociadas a anticuerpos anticitoplasma de neutrófilos (ANCA)
Se define como una lesión patológica con respuesta autoinmune asociada que produce anticuerpos ANCA. Se caracteriza de una vasculitis necrozante con pocos (o nulos) depósitos inmunes, predominantemente afectando a los vasos pequeños(32). Incluyen poliangeitis granulomatosa (GPA o Wegener), poliangeítis microscópica y granulomatosis eosinofílica con poliangeítis (Chug-Strauss o EGPA) y vasculitis renal limitada sin evidencia de vasculitis sistémica. Es caracterizada por ANCA específico para mieloperioxidasa (MPO) o proteinasa-3 (PR3), aunque el 15% de los pacientes con glomerulonefritis pauci-inmune pueden ser negativos para ANCA(17,20).
La histología renal es pauci-inmune (escasez, pero no a la ausencia de depósitos inmunes y de complemento) con glomerulonefritis necrotizante focal y semilunas. La MO presenta necrosis fibrinoide y formación de semilunas(17,20).
La IF no muestra (o poca intensidad) tinción inmunológica. Las semilunas tienen segmentos irregulares o globales de tinción para fibrina en los sitios de necrosis fibrinoide(17).
En la ME muestra pocos o ningún depósito electro-densos principalmente en aquellas con rastros de inmunoglobulina en la IF. Se pueden identificar roturas en la MBG(17).
Las semilunas se definen como una proliferación extracapilar glomerular con 2 o más capas celulares o fibrosas de espesor, se asocian frecuentemente a una presentación de lesión renal aguda rápidamente progresiva(6). Las semilunas se forman por la ruptura en los capilares glomerulares seguido por inflamación, necrosis fibrinoide y proliferación celular; la ruptura de las paredes vasculares libera factores de coagulación que activan la cascada de coagulación y se produce fibrina (necrosis fibrinoide)(6, 32).
La clasificación de Berden ayuda a establecer el grado de actividad patológica y cronicidad de la glomerulonefritis de acuerdo a los hallazgos en microscopía de luz. La clase focal se describe con más del 50% de glomérulos normales, la crescéntica (semilunas) con más del 50% de glomérulos con semilunas celulares, clase mixta con menos del 50% de gomérulos normales, menos del 50% con semilunas y menos del 50% de glomérulos esclerosados y la clase esclerótica con más del 50% de los glomérulos globalmente esclerosados(32).
En las vasculitis pauci-inmune se han encontrado otro tipo de autoanticuerpos diferentes a los ANCAs difícil de confirmar su especificidad. Los anticuerpos anti-lisosomas humanos asociados a protina 2 de membrana (anticuerpos anti-hLAMP2) se han encontrado aproximadamente en el 73% de los pacientes con glomerulonefritis necrotizante pauci-inmune ANCA negativos, igulamente se ha encontrado Pentraxina 3 (PTX3), lo cual puede usarse como diagnóstico para estos pacientes. Los anticuerpos anti-plasminógeno (anti-PLG) se han identificado en las vasculitis con necrosis fibrinoide y en semilunas renales, sin embargo, los estudios que reportan estos hallazgos han utilizado diferentes métodos para la detección de estos anticuerpos. Los anticuerpos anti-moesina se han propuesto en la contribución del daño renal en Japón(31).
Glomerulopatía por anticuerpos anti-membrana basal glomerular
La glomerulopatía con anticuerpos anti-MBG es causada por autoanticuerpos dirigidos contra la cadena alfa3 en el dominio no-colágeno del colágeno tipo 4. Aproximadamente 25% de los pacientes presentan ANCA positivos(17).
Es un tipo de vasculitis esencialmente de vasos pequeños que afecta los capilares glomerulares y capilares alveolares pulmonares. Puede presentarse como una glomerulopatía aislada (glomerulonefritis rápidamente progresiva) o como componente del síndrome riñón-pulmón (síndrome de Goodpasteure)(17).
La MO se caracteriza por necrosis segmentaria a global con formación de semilunas en más del 90% de los pacientes. La tinción PAS y de plata demuestran espacios en la MBG en áreas de necrosis, las áreas sin necrosis pueden mostrar aumento de neutrófilos(17). En la IF se encuentra una tinción intensa en la MBG predominantemente por IgG con C3, que se muestra discontinuo(17).
En la ME no se encuentra ningún tipo de depósitos, se encuentran espacios en la MBG referentes a la necrosis y a la formación de las semilunas. Las semilunas celulares típicamente contienen fibrina electro-densa(17).
En la tabla 1 se muestra un resumen de los nuevos biomarcadores sugeridos y su utilidad para algunas de las glomerulopatías descritas.
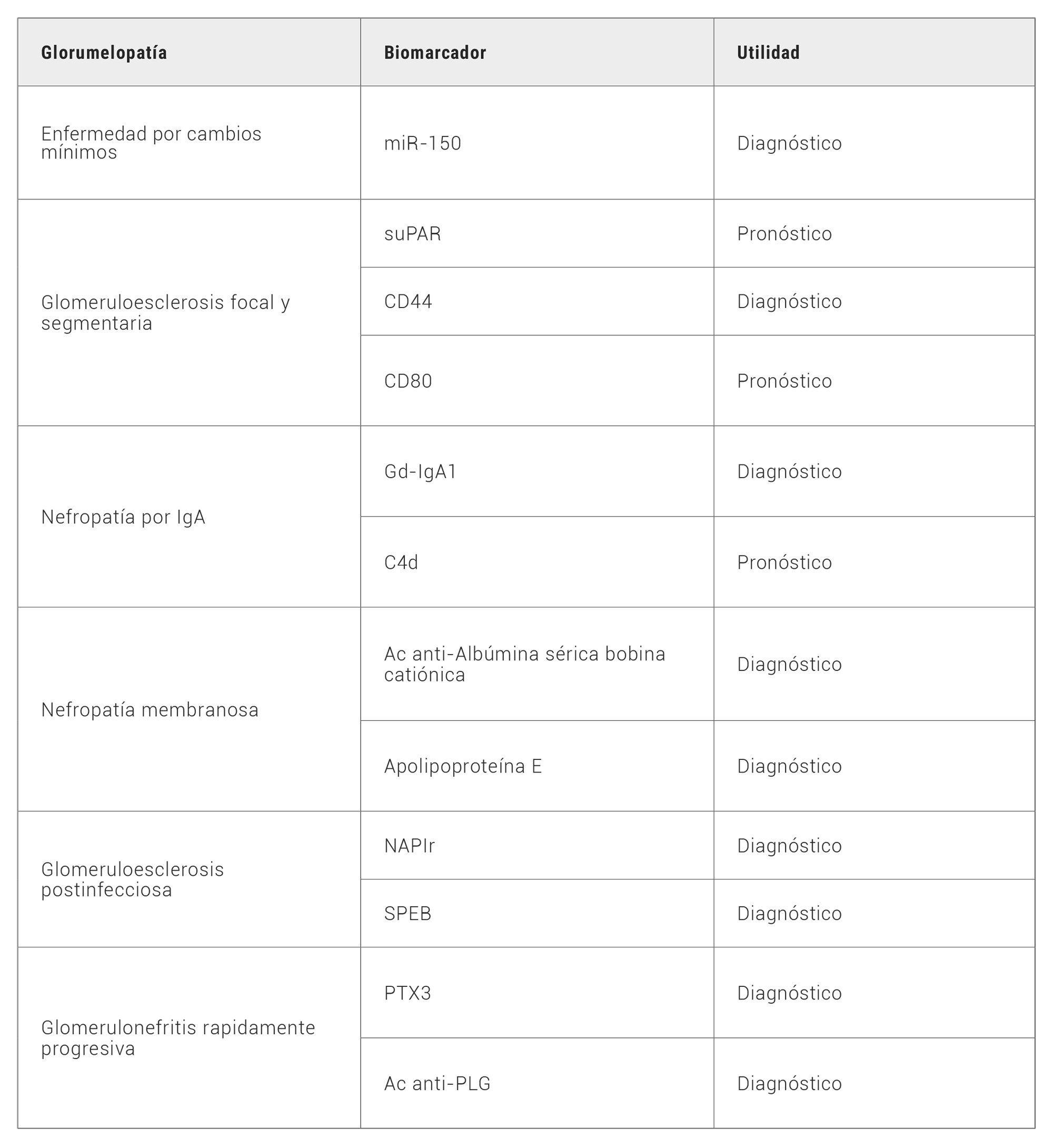
Tabla 1. Nuevos biomarcadores histopatológicos. Ac: anticuerpo
En la tabla 2 se muestra un resumen de las características histopatológicas de cada glomerulopatía primaria descrita.
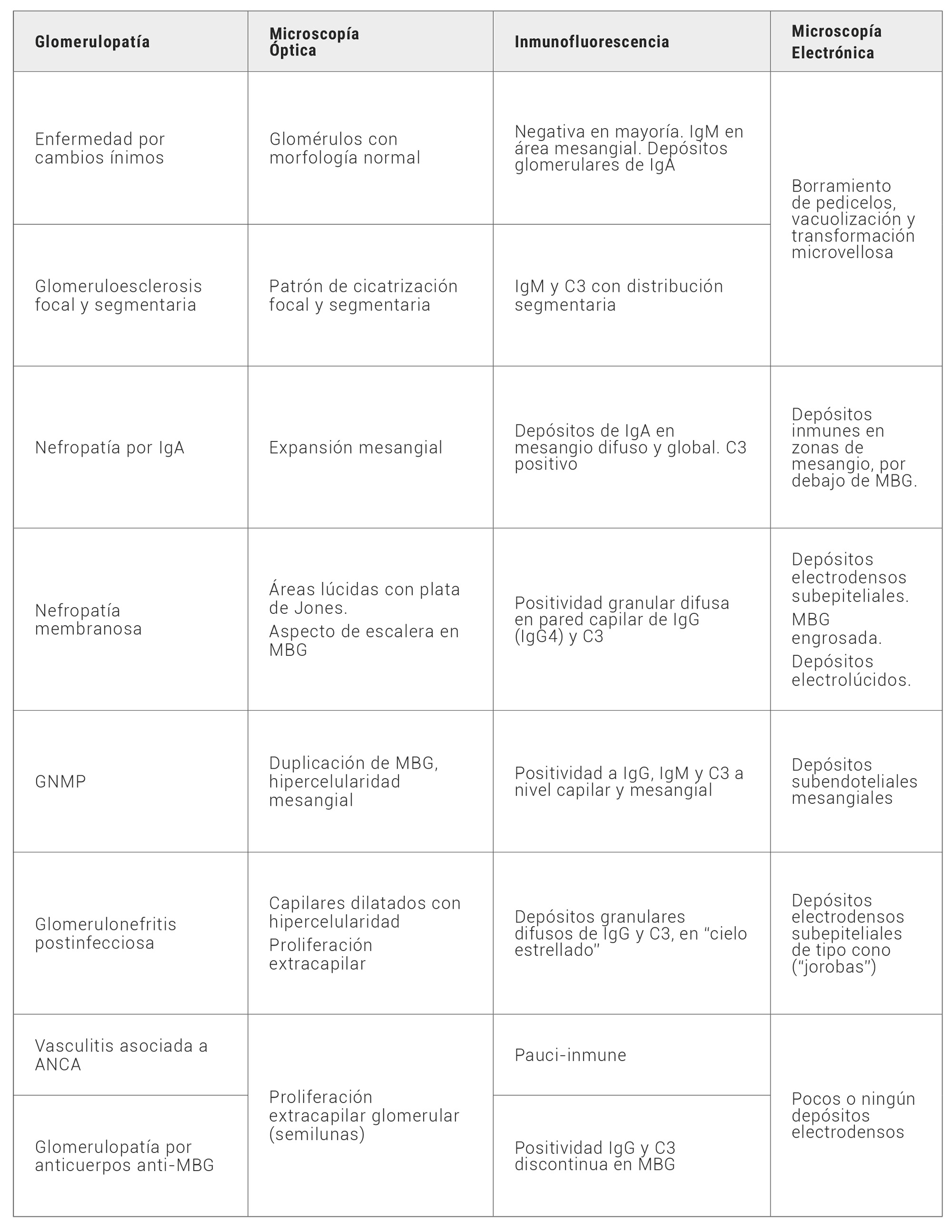
Tabla 2. Patrones histopatológicos de Glomerulopatías primarias. MBG: Membrana basal glomerular; GNMP: Glomerulonefritis membranoproliferativa; ANCA: Anticuerpos anticitoplasma de neutrófilos.
CONCLUSIONES
El diagnóstico específico de las enfermedades glomerulares debe incorporar información relevante de la historia clínica, así como estudios genéticos, histopatológicos, análisis serológicos y urinarios.
La biopsia renal es un método diagnóstico seguro para los pacientes, puede proporcionar importante información para diagnóstico y pronóstico así como puede ser imprescindible para la decisión entre diferentes tratamientos.
En los ensayos clínicos se pueden observar fallos de tratamiento que sugieren variaciones entre individuos en las vías moleculares que impulsan la progresión de la enfermedad, a pesar de una histopatología similar, para poder obtener terapias dirigidas e individualizadas, la identificación de estas vías es necesaria y requerirá un enfoque en los mecanismos operativos a nivel de tejido, en lugar de depender únicamente de los hallazgos histológicos estándar(4).
La presencia de algunos biomarcadores específicos puede ayudar a hacer el diagnóstico diferencial entre patologías de histología parecida. El descubrimiento de biomarcadores no invasivos es necesario para establecer un diagnóstico, pronóstico y metas de tratamiento más específicos con menos riesgo y complicaciones que las que cuenta la biopsia renal percutánea(22). La identificación de ciertos biomarcadores puede presentarse incluso antes de mostrar la enfermedad. La dificultad con estos nuevos biomarcadores es que aún se usan métodos diferentes no estandarizados para su identificación.
AGRADECIMIENTOS
Gracias a la Dra. Rosa Ortega Salas, adscrito del departamento de Anatomía patológica HU Reina Sofía Córdova, por apoyar esta publicación y por haber sido la tutora del trabajo que inspiró este artículo.
REFERENCIAS bibliográficas
- ↑Kumar V., Abbas A.K. & Fausto N., Robbins y Cotran. Patología estructural y funcional. 10a edición. Elsevier. 2019:4.
- ↑Scott E. Wenderfer & Joseph P. Gaut. Glomerular Diseases in Children. Advances in Kidney Disease and Health. 2017 Nov;24(6)364-371. doi:https://doi.org/10.1053/j.ackd.2017.09.005. ScienseDirect 2017 Dec 9.
- ↑Golay, M. Trivedi, A. Abraham, A. Roychowdhary, R. Pandey. The spectrum of glomerular diseases in a single center: A clinicopathological correlation. Indian J Nephrol. 2013 May;23(3):168-175. doi:10.4103/0971-4065.111833. PubMed
- ↑Mark Haas, Surya V. Seshan, Laura Barisoni, Kerstin Amann, Ingeborg M. Bajema, Jan Ulrich Becker, Kensuke Joh, Danica Ljubanovic, Ian S.D. Roberts, Joris J. Roelofs, Sanjeev Sethi, Caihong Zeng, J. Charles Jennette. Consensus Definitions for Glomerular Lesions by Light and Electron Microscopy: Recommendations from a Working Group of the Renal Pathology Society. Kidney International. 2020 Nov;98(5):1120-1134. doi:https://doi.org/10.1016/j.kint.2020.08.006. Elsevier 2020 Aug 28.
- ↑Jurgen Floegel, Sean J. Barbour, Daniel C. Cattran, Jonathan J. Hogan, Patrick H. Nachman, Sydney C.W. Tang, Jack F.M. Wetzels, Michael Cheung, David C. Wheeler, Wolfgang C. Winkelmayer, Brad H. Rovin. Manejo y tratamiento de las enfermedades glomerulares (parte 1): conclusiones de Kidney Disease: Improving Global Outcomes (KDIGO) Controversies Conference. Kidney International. 2019 95:268-280. doi:https://doi.org/10.1016/j.kint.2018.10.018. Elsevier 2019.
- ↑Sheng Nie, Wenjuan He, Ting Huang, Diankun Liu, Guobao Wang, Jian Geng, Nan Chen, Gang Xu, Ping Zhang, Yang Luo, Jing Nie, Xin Xu, Fan Fan Hou. The Spectrum of Biopsy- Proven Glomerular Diseases among Children in China. Clinical Journal of de American Society of Nephrology. 2018 Jul;13(7):1047-1054. doi:10.2215/CJN.11461017. Epub 2018 Jun 18.
- ↑Bandari J, Fuller TW, Turner II RM, D’Agostino LA. Renal biopsy for medical renal disease: indications and contraindications. Review. Can J Urol. 2016 Feb;23(1):8121-8126. PMID:26892051. 2015 Oct.
- ↑Wen Y. Ding, Moin A. Saleem. Current concepts of the podocyte in nephrotic syndrome. Review Article. Kidney Research and Clinical Practice. 2012 Jun;31(2):87-93. doi:https://doi.org/10.1016/j.krcp.2012.04.323. ScienceDirect 2012 Apr 27.
- ↑Cara-fuentes, G., Clapp, W.L., Johnson, R.J. et al. Pathogenesis of proteinuria in idiopathic minimal change disease: molecular mechanisms. Pediatr Nephrol. 2016 31:2179-2189. doi:https://doi.org/10.1007/s00467-016-3379-4. Springer Link 2016 Jul 06
- ↑Hashmi MS, Pandey J. Nephritic Syndrome. StatPearls Publishing, Treasure Island (FL) 2021 Sept. PMID:32965911. Europe PMC Aug 2022.
- ↑Taheri S. Renal biopsy reports in nephritic syndrome: Update. World Journal of Nephrology. 2022 Mar;11(2):73-85. doi:10.5527/wjn.v11.i2.73. PubMed 25 Mar 2022.
- ↑Priscilla Kincaid-Smith & Kenneth Fairley. The Investigation of Hematuria. Seminars in Nephrology. 2005 May;25(3):127-135. doi:https://doi.org/10.1016/j.semnephrol.2005.01.002. Elsevier 2008 Sept 21.
- ↑Sara Bielsa-García, Ana Garzarán, Celia del Agua, Antonio Gascón, José-Esteban Ruiz, Francisca Berisa. Glomerulonefritis rápidamente progresiva: a propósito de dos casos. NefroPlus. 2016 Dec;8(2):170-178. doi:Nefrología 2016.
- ↑Arimura, Y., Muso, E., Fujimoto, S. et al. Evidence-based clinical practice guidelines for rapidly progressive glomerulonephritis 2014. Clin Exp Nephrol. 2016 Jun;20:322-341. doi:https://doi.org/10.1007/s10157-015-1218-8.
- ↑Marina Vivarelli, Laura Massella, Barbara Ruggiero, Emma Francesco. Minimal Change Disease. Clin J Am Soc Nephrol. 2017 12(2):332-345. doi:10.2215/CJN.05000516. PubMed 2016 Dec 9.
- ↑R Bertelli, A Bonanni, A Di Donato, M Cioni, P Ravani, G M Ghiggeri. Regulatory T cells and minimal change nephropathy: in the midst of a complex network. Clinical & Experimental Inmunology. 2015 Feb;183(2):166-174. doi:https://doi.org/10.1111/cei.12675. Europe PMC 2015 Oct 12.
- ↑Fogo A., Kashgarian M. Atlas Diagnóstico de patología renal. Editorial Elsevier Saunders S.A. 3ª ed. 2018:13-119.
- ↑Froes, B.P., de Almeida Araújo, S., Bambirra, E.A. et.al. Is CD44 in glomerular parietal epithelial cells a pathological marker of renal function deterioration in primary focal segmental glomerulosclerosis?. Pediatr Nephrol. 2017 32:2165-2169. doi:https://doi.org/10.1007/s00467-017-3775-4. Springer Link 2017 Aug 10.
- ↑Ishizuka K., Miura K. Hashimoto T., Kaneko N., Harita Y., Yabuuchi T., Hisano M., Fujinaga S., Omori T., Yamaguchi Y., Hattori M. Degree of foot process effacement in patients with genetic focal segmental glomerulosclerosis: a single-center analysis and review of literature. Scientific Reports. 2021 Jun;11(1):12008. doi:10.1038/s41598-021-91520-9. Nature 2021 Jun 08.
- ↑Brad H. Rovin, Dawn J. Caster, Daniel C. Cattran, Keisha L. Gibson, Jonathan J. Hogan, Marcus J. Moeller, Dario Roccatello, Michael Cheung, David C. Wheeler, Wolfgang C. Winkelmayer and Jürgen Floegel. Manejo y tratamiento de las enfermedades glomerulares (parte 2): conclusiones de Kidney Disease: Improving Global Outcomes (KDIGO) Controversies Conference. Kidney International 2019 95:281-295. doi:https://doi.org/10.1016/j.kint.2018.11.008. Elsevier 2019.
- ↑Meling Lu, Cheng Wang, Yunlong Yuan, Yuanyuan Zhu, Zhimin Yin, Zhengkun Xia, Chunni Zhang. Differentially expressed microRNAs in kidney biopsies from various subtypes of nephrotic children. Experimental and Molecular Pathology. 2015 Dec;99(3):590-595. doi:https://doi.org/10.1016/j.yexmp.2015.10.003. Elsevier 2015 Oct 19.
- ↑Cara-Fuentes, G., Venkatareddy, M., Verma, R. et al. Glomerular endothelial cells and podocytes can express CD80 in patients with minimal change disease during relapse. Pediatr Nephrol. 2020;35:1887-1896. doi:https://doi.org/10.1007/s00467-020-04541-3. Springer Link 2020 May 12
- ↑Suzuki H., Biomarkers for IgA nephropathy on the basis of multi-hit pathogenesis. Clin Exp Nephrol. 2019;23:26-31. doi:https://doi.org/10.1007/s10157-018-1582-2. Springer Link 2018 May 08.
- ↑Alfons Segarra, Katheryne Romero, Irene Agraz, Natalia Ramos, Alvaro Madrid, Clara Carnicer, Elias Jatem, Ramón Vilalta, Luis Enrique Lara, Elena Ostos, Naiara Valtierra, Juliana Jaramillo, Karla V. Arredondo, Gema Ariceta, & Cristina Martinez. Mesangial C4d deposits in early IgA nephropathy. Clin J Am Soc Nephrol. 2018 Feb;13(2):258-264. doi:10.2215/CJN.02530317. PubMed 2017 Nov 16.
- ↑Ronco P., Debiec H. Molecular Pathogenesis of Membranous Nephropathy. Annual review of Pathology. 2019 Oct;15:287-313. doi:10.1146/annurev-pathol-020117-043811. AR 17 Oct 2019.
- ↑Akihiko Koshino, Chikako Takaeda, Takahiro Matsuno, Shinji Kitajima, Yasunori Iwata, Norihiko Sakai, Kiyotaka Nagahama, Yo Niida, Takao Saito, Hitoshi Yokoyama & Takashi Wada. Membranous nephropathy-like Apolipoprotein E deposition disease with Apolipoprotein E Toyonaka and Homozygous Apolipoprotein E2/2 without dislipidemia, with characteristic electron-dense deposits. Case Rep Nephrol Dial. 2022 Aug;12(2):96-104. doi:10.1159/000525086. Karger 10 Jun 2022.
- ↑Andreia Marinhas, Telma Luís, Caatarina Neves, Carmen Carmo & Clara Gomes. Membranoproliferative glomerulonephritis: A rare pediatric nephropathy. Port J Nephrol Hypert. 2022;36(1):7-10. doi:http://doi.org/10.32932/pjnh.2022.03.175. Publicacoes Ciencia & Vida 2022 Mar 21.
- ↑Masani, Naveed; Jhaveri, Kenar D. & Fishbane, Steven. Update on Membranoproliferative GN. Clin J Am Soc Nephrol. 2014 Mar;9(3):600-608. doi:10.2215/CJN.06410613. PubMed 2014 Jan 9.
- ↑Kambham, Neeraja MD. Postinfectious Glomerulonephritis. Adv Anat Pathol. 2012 Sept;19(5):338-347. doi:10.1097/PAP.0b013e31826663d9. PMID:22885383.
- ↑Chauvet Sophie, Berthaud, Romain; Devriese, Magali; Mignotet, Morgane; Vieira Martins, Paula; Robe-Rybkine, Tania; Miteva, Maria A.; Gyulkhandanyan, Aram; Ryckewaert, Amélie; Louillet, Ferielle; Merieau, Elodie; Mestrallet, Guillaume; Rousset-Rouvière, Caroline; Thervet, Eric; Hogan, Julien; Ulinski, Tim; Villoutreix, Bruno O.; Roumenina, Lubka; Boyer, Olivia & Frémeaux-Bacchi, Véronique. Anti-Factor B antibodies and acute postinfectious GN in children. JASN 2020 Apr;31(4):829-840. doi:10.1681/ASN.2019080851. Epub 2020 Feb 7.
- ↑Tadesco, M., Gallieni, M., Pellegata, F. et al. Update on ANCA-associated vasculitis: from biomarkers to therapy. J Nephrol. 2019 Dec;32:871-882. doi:https://doi.org/10.1007/s40620-019-00628-9. Springer Link 2019 Jul 12.
- ↑Jennette, J. Charles & Nachman, Patrick H. ANCA Glomerulonephritis and Vasculitis. Clin J Am Soc Nephrol. 2017 Oct;12(10):1680-1691. doi:10.2215/CJN.02500317. Europe PMC 2017 Aug 25.
INFORMACIÓN DEL ARTÍCULO
Conflicto de intereses: Los autores/as de este artículo declaran no tener ningún tipo de conflicto de intereses respecto a lo expuesto en el presente trabajo.
Correspondencia: Zyanya Gabriela Tovar Torres. Hospital Infantil de México “Federico Gómez”. Calle Dr Márquez 162 Delegación:, Doctores, Cuauhtémoc, 06720 Ciudad de México, CDMX, México. E-mail: diana_caroli_93@hotmail.com