

Caso Clínico
Implicación del laboratorio en el diagnóstico y seguimiento de la hemoglobinuria paroxística nocturna: presentación de un caso clínico
Laboratory involvement in the diagnosis and monitoring of paroxysmal nocturnal hemoglobinuria: a case report
Actual Med. 2021; 106(812): 89-92 DOI: 10.15568/am.2021.812.cc04
Recibido: 25/01/2020
Revisado: 20/04/2020
Aceptado: 25/04/2020
Resumen
La hemoglobinuria paroxística nocturna es una rara enfermedad clonal no maligna, causada por una mutación somática adquirida en un número variable de células madres hematopoyéticas, cuya consecuencia es una sensibilidad anormal de las células sanguíneas a la lisis mediada por el complemento. Se manifiesta con anemia hemolítica intravascular, un grado variable de insuficiencia medular y elevado riesgo trombótico. El manejo de la enfermedad se basa en un adecuado diagnóstico y seguimiento clínico y de laboratorio. El método de elección en el diagnóstico y monitorización del paciente es la citometría de flujo. El único tratamiento curativo es el trasplante de células madres hematopoyéticas. Eculizumab es el primer tratamiento especifico aprobado para esta enfermedad.
Presentamos el caso de una paciente diagnosticada de hemoglobinuria paroxística nocturna, que recientemente ha iniciado tratamiento con eculizumab. Mostramos la evolución clínica y analítica durante los seis primeros meses de tratamiento como valoración del beneficio del mismo.
Palabras clave: Hemoglobinuria paroxística nocturna; Citometria de flujo; Eculizumab.
Abstract
Paroxysmal nocturnal hemoglobinuria is a rare non malignant clonal disease, caused by an acquired somatic mutation in a variable number of hematopoietic stem cells, whose consequence is an abnormal sensitivity of blood cells to complement-mediated lysis. It manifests as intravascular hemolytic anemia, a variable degree of bone marrow insufficiency and high thrombotic risk. The effective management of the disease is based on an adequate diagnosis and clinical and laboratory follow-up. Flow cytometry is the method of choice for diagnosis and monitoring of the patient. The only curative treatment is hematopoietic stem cell transplantation. Eculizumab is the first specific treatment approved for this disease.
We present the case of a patient diagnosed with paroxysmal nocturnal hemoglobinuria, wo has recently started treatment with eculizumab. We show the clinical and analytical evolution during the first months of treatment as an assessment of is benefit.
Keywords: Paroxysmal nocturnal hemoglobinuria; Flow cytometry; Eculizumab.
Leer Artículo Completo
INTRODUCCIÓN
La hemoglobinuria paroxística nocturna (HPN) es un trastorno clonal adquirido de las células madres hematopoyéticas. Está causado por una mutación somática del gen PIG-A (Glucosil-Fosfatidil-Inositol A), localizado en el brazo corto del cromosoma X (1).
Se trata de una enfermedad rara, con una incidencia estimada de 0,05-0,13 casos por 100.000 habitantes/año (2). Aunque se puede manifestar a cualquier edad, la incidencia es mayor en la tercera década de la vida, afectando por igual a ambos sexos. Presenta una elevada mortalidad, con una mediana de supervivencia de 10-15 años tras el diagnóstico (3).
Las células afectadas por la mutación PIG-A son deficientes o carentes en proteínas del grupo de anclaje
glicosilfosfatidilinositol (GPI), necesario para la unión de numerosas proteínas a la membrana de las células sanguíneas. Dos de estas proteínas, el factor acelerador de la degradación del complemento (DAF) y el inhibidor de la lisis reactiva de la membrana (ILRM), son inhibidores fisiológicos del complemento, por lo que en estos pacientes los hematíes presentan una mayor sensibilidad a la lisis mediada por el complemento (4).
Clínicamente se manifiesta con anemia hemolítica crónica con crisis de reagudización paroxísticas, un grado variable de insuficiencia medular y predisposición a sufrir procesos trombóticos, no siendo la hemoglobinuria una manifestación frecuente. Las complicaciones de la enfermedad son consecuencia directa de la hemólisis intravascular, cuya gravedad refleja el tamaño del clon HPN, y de la fijación de óxido nítrico por la hemoglobina plasmática libre. El déficit de óxido nítrico va a provocar vasoconstricción, disfunción plaquetaria, endotelial y de la musculatura lisa, manifestándose con dolor abdominal, torácico y lumbar, espasmos esofágicos, disfagia, disfunción eréctil, enfermedad renal, astenia intensa y elevado riesgo trombótico. La insuficiencia renal, tanto aguda como crónica, y la hipertensión pulmonar son complicaciones especialmente graves de la enfermedad (3).
La complicación más frecuente, y principal causa de muerte, es la trombosis, cuyo riesgo está directamente relacionado con el tamaño del clon HPN. En la mayoría de casos se producen en localizaciones inusuales del territorio venoso, aunque también pueden producirse trombosis arteriales (4,5).
Existe una asociación bien establecida entre HPN e insuficiencia medular: un 30-45% de pacientes con HPN presentan cierto grado de insuficiencia medular, y un 40-70% de pacientes con aplasia medular alberga un clon HPN detectable. Estos últimos pacientes van a responder mejor a la terapia inmunosupresora, pero tienen riesgo de sufrir una expansión clonal durante la recuperación de la aplasia y desarrollar una HPN manifiesta (6).
El manejo adecuado de esta enfermedad se basa en un diagnóstico preciso y en el seguimiento tanto clínico como de laboratorio (7). Los estudios de laboratorio muestran un grado variable de anemia, con datos de hemólisis intravascular (hiperbilirrubinemia indirecta, elevación de la enzima láctico deshidrogenasa-LDH- y descenso de haptoglobina,) y test de Coombs directo negativo. Otros posibles hallazgos son hemoglobinuria, ferropenia, leucopenia y trombopenia, y en los casos en los que no exista aplasia medular asociada reticulocitosis (2).
La citometría de flujo es el método de referencia para identificar los clones HPN. Se trata de una técnica sensible y especifica que proporciona un análisis cuantitativo y cualitativo de proteínas ancladas a GPI en las células sanguíneas, lo que permite tanto el diagnóstico como la monitorización de la enfermedad y del tratamiento, incluso en pacientes con clones HPN pequeños (3,7). El estudio se realiza rutinariamente en muestras de sangre periférica, en al menos dos líneas celulares, empleando dos anticuerpos monoclonales diferentes. En un primer paso se analizan los granulocitos neutrófilos y monocitos, y en caso de detectarse un déficit de proteínas ancladas mediante GPI se analizan los hematíes, ya que el análisis aislado de hematíes puede dar falsos negativos en caso de crisis hemolítica o transfusión recientes (2,3).
El único tratamiento curativo de enfermedad es el trasplante de células madres hematopoyéticas. Está indicado como tratamiento de primera línea solo en pacientes que asocien una insuficiencia medular grave debido a su elevada morbimortalidad (8).
El primer tratamiento especifico para la HPN aprobado por la FDA (Agencia Americana del Medicamento) y la EMA (Agencia Europea del Medicamento) es eculizumab. Se trata de un anticuerpo monoclonal Ig G que actúa uniéndose selectivamente a la proteína C5 del complemento, impidiendo la formación del complejo de ataque a la membrana y reduciendo la hemólisis intravascular, aunque sin interferir en la hemolisis extravascular, ya que no actúa sobre la fracción C3 del complemento. Su uso estabiliza los niveles de hemoglobina, reduce las necesidades transfusionales, disminuye el riesgo trombótico, mejora la función renal, la hipertensión pulmonar, la astenia, la calidad de vida y supervivencia global de estos pacientes. Por su mecanismo de acción, eculizumab aumenta el riesgo de infección por bacterias encapsuladas, especialmente Neisserias (5,8).
CASO CLÍNICO
Paciente con antecedentes de aplasia medular grave adquirida, diagnosticada a los 19 años de edad. La citometría de flujo no detectó clones HPN en el momento del diagnóstico. El trasplante de células madres hematopoyéticas no fue posible ante la ausencia de un donante HLA compatible. Recibió soporte transfusional y tratamiento inmunosupresor con ciclosporina, con escasa respuesta, presentando varios episodios de sangrado de diversos orígenes relacionados con trombopenias intensas que requirieron ingreso hospitalario. Debido a la falta de respuesta a la inmunosupresión, se inició tratamiento con andrógenos (danazol) y corticoides (prednisona), obteniéndose una respuesta completa, con reducción importante de los requerimientos transfusionales, infecciones intercurrentes y episodios de sangrado . Tres años más tarde presentó un cuadro de dolor abdominal, ictericia y orinas oscuras. El estudio de laboratorio puso de manifiesto datos de hemólisis intravascular, con test de Coombs directo negativo y hemoglobinuria. La citometría de flujo detectó clones HPN III (con deficiencia total de proteínas ancladas a GPI) en monocitos (93%), granulocitos (94%) y hematíes (73%). Desde entonces presenta crisis hemolíticas esporádicas, que en la mayoría de ocasiones han requerido soporte transfusional. En el último año ha sufrido un aumento importante en la incidencia de episodios hemolíticos, con mayores requerimientos transfusionales y astenia intensa, por lo que se indicó tratamiento con eculizumab. La pauta de tratamiento inicial establecida ha sido de 600 mg semanales durante 4 semanas, seguida de una pauta de mantenimiento de 900 mg cada dos semanas.
Siguiendo las recomendaciones del Consenso español para el diagnóstico y tratamiento de la HPN, previamente recibió vacunación frente a Neisseria meningitidis (B y C, y combinada A, C, Y, W-135) y profilaxis con penicilina, lo que desencadenó una nueva una crisis hemolítica.
En la figura 1 se muestra la evolución analítica de la paciente desde el inicio del tratamiento.
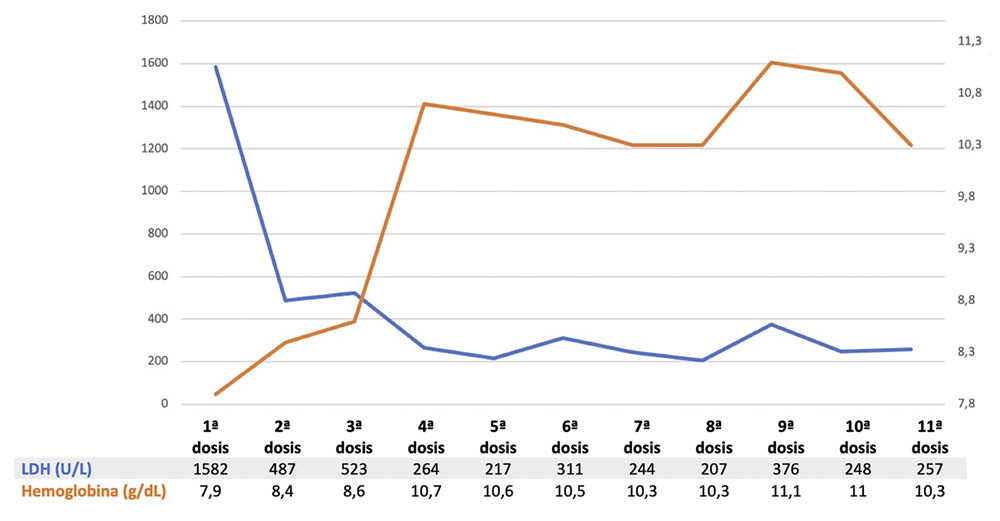
Figura 1. Evolución de la enzima láctico deshidrogenasa (LDH) y de las concentraciones de hemoglobina durante los seis primeros meses de tratamiento con eculizumab. Como se observa, con la primera dosis de eculizumab se ha producido un descenso notable de la concentración sérica de LDH, que se ha mantenido durante el seguimiento. Los niveles de hemoglobina se han estabilizado, aunque solo se han recuperado ligeramente y la anemia ha persistido.
DISCUSIÓN
La hemoglobinuria paroxística nocturna es el único trastorno hemolítico adquirido causado por una mutación somática en un gen. Es una enfermedad rara, de gran variabilidad clínica y difícil diagnóstico, que asocia una elevada morbimortalidad.
Aunque la principal manifestación clínica es la anemia hemolítica intravascular, se puede presentar como una insuficiencia medular o una trombosis, siendo el dolor torácico, abdominal y lumbar, la astenia intensa, disnea, disfagia, disfunción eréctil y la insuficiencia renal otras posibles manifestaciones. Esta gran variabilidad clínica hace que pase inadvertida, si no es considerada como un posible diagnóstico, o que se diagnostique tardíamente. Actualmente existe un tratamiento específico y eficaz para la enfermedad, por lo que el diagnóstico precoz supone una importante mejora en la calidad de vida y pronóstico de estos pacientes. Eculizumab es un anticuerpo monoclonal humanizado que bloque la formación del complejo de ataque a la membrana, reduciendo eficazmente la hemólisis intravascular. Recientemente la EMA ha modificado los criterios de indicación del anticuerpo, incluyendo a pacientes con alta actividad de enfermedad, definida por hemólisis elevada junto a uno o mas síntomas asociados como astenia, dolor abdominal, hemoglobinuria, disnea, anemia, trombosis, disfagia y/o disfunción eréctil, independientemente de las necesidades transfusionales.
En el caso que presentamos, la paciente presentaba frecuentes crisis hemolíticas, con cifras de LDH>1,5xLSN y astenia intensa, por lo que se indicó eculizumab. Siguiendo las recomendaciones del Consenso español para el diagnóstico y tratamiento de la HPN, hemos revisado el beneficio del tratamiento durante los seis primeros meses.
Durante este periodo, la tolerancia a eculizumab ha sido adecuada y la paciente ha experimentado una mejoría clínica significativa, con reducción importante de la astenia, probablemente relacionado con una recuperación de los niveles de óxido nítrico, ya que la anemia ha persistido. No ha presentado ninguna crisis hemolítica, complicación de gravedad ni ha requerido soporte transfusional.
Desde el punto de vista analítico, se ha producido un descenso notable de la concentración sérica de LDH, como reflejo de la reducción de la hemólisis intravascular. Los niveles de hemoglobina se han estabilizado, aunque la recuperación de la hemoglobina ha sido ligera y la anemia ha persistido, probablemente debido a un componente de hemólisis extravascular sobre el que no actúa el anticuerpo eculizumab.
Siguiendo las recomendaciones del Consenso Español para diagnóstico y tratamiento de la HPN, se indica continuar con el tratamiento dada la mejoría clínica y analítica de la paciente.
REFERENCIAS BIBLIOGRÁFICAS
- ↑ Mon Père N, Lenaerts T, Pacheco JM, Dingli D. Evolutionary dynamics of paroxysmal nocturnal hemoglobinuria. PLoS Comput Biol. 2018;14(6):e1006133. DOI: 10.1371/journal.pcbi.1006133
- ↑ Macía I, Garcia T, Fundora T, Fernández N. Paroxysmal nocturnal hemoglobinuria: from Strubing to Eculizumab. Rev Cubana Hematol Inmunol Hemoter. 2013;29(3):218-232
- ↑Villegas A, Arrizabalaga B, Bonanad S et al. Spanish consensus statement for diagnosis and treatment of paroxysmal nocturnal haemoglobinuria. Med Clin (Barc). 2016;146(6):239-286. DOI: 10.1016/j.medcli.2015.12.012
- ↑ Elias NS, Riedl J, Stouten K, Levin MD et al. Abdominal pain in combination with an unexplained hemolytic anemia are crucial signs test for paroxysmal nocturnal hemoglobinuria: A case report. Clin Case Rep. 2019;7(1):175-179. DOI: 10.1002/ccr3.1771
- ↑ Wong EK, Kavanagh D. Diseases of complement dysregulation: an overview. Semin Immunopathol. 2018;40(1):49-64. DOI: 10.4081/hr.2017.7012
- ↑Manivannan P, Ahuja A, Pati HP. Diagnosis of paroxysmal nocturnal hemoglobinuria: Recent advances. Indian J Hematol Blood Transfus, 2017; 33(4):453-462. DOI:10.1007/s12288-017-0868
- ↑ Correia RP, Bento LC, Bortolucci AC et al. Technical advances in flow cytometry based diagnosis and monitoring of paroxysmal nocturnal hemoglobinuria. Einstein (Sao Paulo). 2016;14(3):366-373. DOI: 10.1590/S1679-45082016AO3641
- ↑ Macedo ES, Parente SLA, Pro JDZ et al. Renal involvement in paroxysmal nocturnal hemoglobinuria: a brief review of the literature. Rev Assoc Med Bras. 2018;64(12):1139-1146. DOI: 10.1590/1806-9282.64.12.1139
INFORMACIÓN DEL ARTÍCULO
Conflicto de intereses: Los autores/as de este artículo declaran no tener ningún tipo de conflicto de intereses respecto a lo expuesto en el presente trabajo.
Autora para la correspondencia: María José Ruíz Márquez. Hospital de Riotinto. Avda de la Esquila, 5. 21660 · Minas de Riotinto, Huelva. E-mail: ruizmarquezmj@gmail.com





