

Cartas al Editor
Hemorragia vítrea unilateral como primera manifestación del síndrome de von Hippel-Lindau
Unilateral vitreous hemorrhage as first manifestation in von Hippel-Lindau syndrome
Actual. Med. 2015; 100: (794): 47-48 DOI: 10.15568/am.2015.794.cd01
Enviado: 29-12-2014
Revisado: 26-01-2015
Aceptado: 9-02-2015
Leer Artículo Completo
Estimado editor:
El síndrome de von-Hippel-Lindau es una enfermedad de herencia autosómica dominante debida a una mutación en el cromosoma 3 si bien existe hasta un 20% de mutaciones “de novo”.
Se caracteriza por el desarrollo de tumores benignos y malignos en la retina y el cerebro, junto con quistes y tumores en varios órganos viscerales como el páncreas, glándulas suprarrenales y órganos reproductivos (1).
Las manifestaciones oculares suelen ser las primeras en diagnosticarse.
Las más frecuentes son hemangioblastomas capilares retinianos de localización periférica o yuxtapapilar (menos frecuente) (1).
Se suelen mantener asintomáticos hasta la tercera década de la vida, cuando aparece disminución de la agudeza visual por hemorragia y exudación en la vecindad del tumor.
Dejadas a su evolución pueden provocar desprendimiento de retina, neovascularización, glaucoma y ptisis bulbi.
Existen varias opciones de tratamiento dependiendo del tamaño y localización del tumor :fotocoagulación con láser, crioterapia, terapia fotodinámica, cirugía vitreoretinal y antiangiogénicos (2).
Presentamos el caso de un paciente varón de 25 años sin antecedentes personales ni familiares de interés, que acude a urgencias por pérdida de visión de forma brusca en ojo derecho, detectándose en la exploración una hemorragia vítrea que impide visualizar la retina, siendo la exploración del ojo izquierdo normal.
Tras la resolución de la hemorragia se observan en total seis angiomas en la retina, con exudación que llega a mácula.
Se solicitan pruebas de imagen y se detectan lesiones quísticas en varios órganos viscerales.
Tras realizar el estudio sistémico el paciente es diagnosticado de síndrome de von Hippel Lindau y cuando la hemorragia vítrea lo permite se inicia la fotocoagulación con láser de los angiomas (figura 1).
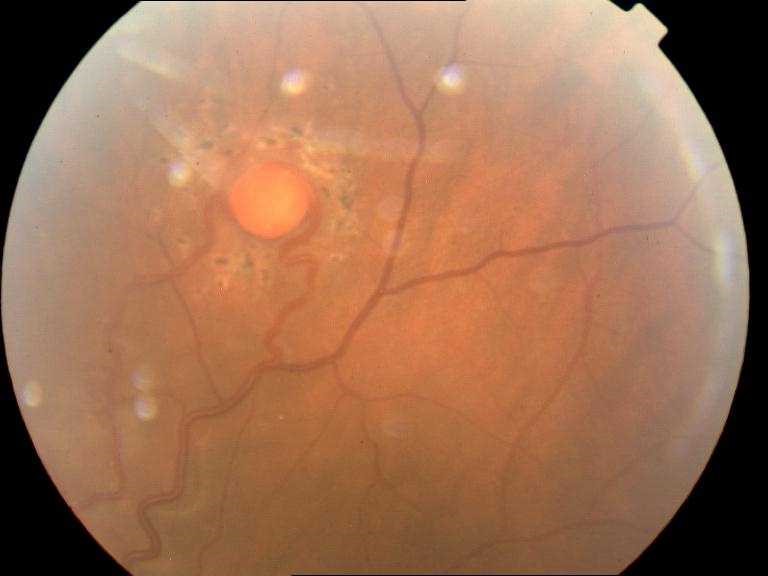
Figura 1. Angioma fotocoagulado.
Durante su evolución el ojo derecho alcanza una agudeza visual de la unidad y la exudación se va reabsorbiendo.
La detección de estas lesiones oculares, incluso como única manifestación de la enfermedad y sin historia familiar nos debe hacer tener en cuenta el diagnóstico y se deben explorar todos los órganos que pueden estar afectados, así como realizar test genéticos a los familiares (3).
Los pacientes afectados y los familiares que presenten la mutación deberán someterse a revisiones periódicas durante toda su vida (3).
Por lo general las lesiones aumentan de tamaño progresivamente y rara vez regresan, por lo que es recomendable el tratamiento profiláctico cuando aún son pequeñas.
No existe un tratamiento estándar para los hemangiomas retinianos, la elección depende principalmente del tamaño de la lesión, localización y complicaciones asociadas (4).
Los pacientes deben ser sometidos de forma periódica a exploraciones neurológicas, fondo de ojo y pruebas de imagen abdominal y cerebral. De esta forma se pueden atenuar o evitar las consecuencias de la enfermedad, tanto oculares como sistémicas.
BIBLIOGRAFÍA
- Staub B., Livingston AD, Chévez-Barrios P, et al. Hemangioblastoma of the optic nerve producing bilateral optic tract edema in a patient with von Hippel–Lindau disease. 2014; 5: 33
- Ammerman JM, Lonser RR, Dambrosia J, Butman JA, Oldfield EH. Long-term natural history of hemangioblastomas in patients with von hippel-lindau disease: Implications for treatment. J Neurosurg. 2006;105:248–55
- Haddad NM, Cavallerano JD, Silva PS. Von hippel-lindau disease: A genetic and clinical review. Semin Ophthalmol. 2013;28:377–86
- Chan CC, Collins AB, Chew EY. Molecular pathology of eyes with von hippel-lindau (VHL) disease: A review. Retina. 2007;27:1–7
INFORMACIÓN DEL ARTÍCULO
Autora para la correspondencia: Ana María Alfaro Juárez. alfaro.juarez@hotmail.com





