

Cartas al Editor
Amiloidosis cardiaca y macroglobulinemia de Waldenström
Cardiac amyloidosis and Waldenström macroglobulinemia
Actual. Med. 2016; 101: (798): 136-137 DOI: 10.15568/am.2016.798.cd03
Enviado: 14-07-2016
Revisado: 30-07-2016
Aceptado: 20-08-2016
Leer Artículo Completo
Estimado Editor,
Se presenta el caso de una mujer de 51 años diagnosticada dos años antes de macroglobulinemia de Waldenström (MW) por presentar astenia progresiva de dos años de evolución asociada a pérdida de 10 Kg de peso, cefalea, mareo, visión borrosa, anemia moderada, VSG en la primera hora de 95 mm, hepatoesplenomegalia, adenopatías laterocervicales y retroperitoneales, polineuropatía sensitivo-motora distal, e IgM de 4396 mg/dL, asociado a un inmunofenotipo en médula ósea compatible. Tras ser valorada por Hematología y ante la exacerbación de la clínica en los meses siguientes al diagnóstico, se inició tratamiento con bendamustina-rituximab con buena respuesta clínica, estando en seguimiento estrecho por su parte.
En los últimos seis meses presenta palpitaciones y mareo en el contexto de hipotensiones arteriales, así como en la última semana disnea progresiva con ortopnea y edematización periférica, agravadas en las horas previas a su consulta por taquicardia supraventricular a 195 lpm, motivo por el que se decide ingreso hospitalario para tratamiento y estudio. Se realiza ecocardiografía transtorácica con datos sugerentes de amiloidosis cardiaca, resultado confirmado posteriormente por angio-RM cardiaca (Figura 1) y biopsia de grasa subcutánea, identificando el subtipo de amiloidosis AL. Tras valoración multidisciplinar, se descartó que la paciente fuese candidata a trasplante cardiaco ni a quimioterapia paliativa, instaurándose exclusivamente el tratamiento propio de la disfunción cardiaca. Tras una tórpida evolución, la paciente falleció 9 meses tras el diagnóstico.
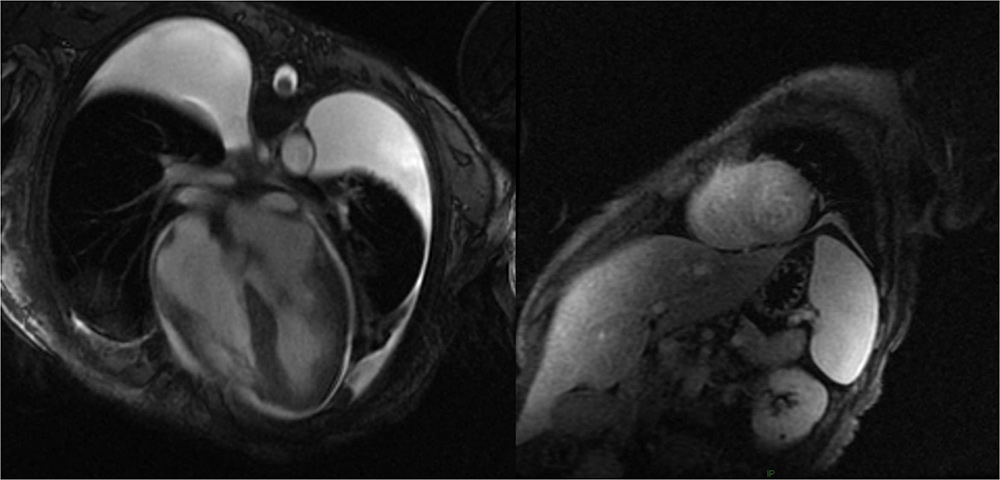
Figura1
El diagnóstico final fue de Amiloidosis cardiaca como complicación de una macroglobulinemia de Waldenström.
La MW es una paraproteinemia que consiste en la proliferación monoclonal de células linfoides B secretoras de IgM, que suele manifestarse con síntomas derivados de la infiltración linfoplasmocitaria, siendo también muy típica la aparición de un síndrome de hiperviscosidad asociado la estructura pentamérica de la IgM (diátesis hemorrágica, trastornos cardiovasculares, neurológicos y visuales). Suele afectar a varones (70%) de edad avanzada, presentando una incidencia de 0.5 casos nuevos por cada 100000 habitantes y año. La MW es habitualmente una enfermedad muy indolente, con una mediana de supervivencia superior a los 10 años desde el diagnóstico, que sólo precisa tratamiento en pacientes sintomáticos o con datos de progresión de la enfermedad(1,2).
Las discrasias de células plasmáticas, fundamentalmente el mieloma múltiple (10-15%) y con menor frecuencia las gammapatías monoclonales IgM (2.2%), pueden asociar como complicación durante su evolución una amiloidosis, siendo el tipo más frecuente la amiloidosis AL, aunque hay algunos casos también de AA(3-5). La amiloidosis primaria o AL es una enfermedad poco común (5.1 a 12.8 casos nuevos por millón de habitantes y año) que consiste en el depósito tisular de fibrillas proteicas insolubles compuestas por fragmentos de una cadena ligera (kappa o lambda) monoclonal en disposición beta plegada(6), y que se tiñe con colorante rojo Congo dando lugar a una patognomónica birrefringencia verde manzana al observarse bajo luz polarizada(4,5). Las manifestaciones clínicas dependen de dónde se produzca el depósito de amiloide, siendo los órganos más frecuentemente afectados riñón (53-74%), corazón (35-60%), ganglios linfáticos (21%), hígado (14-27%), sistema nervioso periférico (3-22%) y sistema nervioso autónomo (7-18%), presentando en el momento del diagnóstico un compromiso de más de un órgano el 54-69% de los pacientes(4-6).
La amiloidosis cardiaca aparece hasta en el 60% de los pacientes con amiloidosis AL y sólo en 5% de los pacientes con amiloidosis AA, empeorando de forma marcada el pronóstico principalmente el primer grupo (mediana de supervivencia de 11-12 meses desde el diagnóstico)(3-5,7). Los síntomas con los que se manifiesta con más frecuencia la amiloidosis cardiaca son: insuficiencia cardiaca, presíncopes o síncopes, alteraciones en el sistema de conducción o afectación pericárdica(5-8). En cualquier caso el diagnóstico de amiloidosis cardiaca se basa en la realización de una ecocardiografía bidimensional, pudiendo completarse el estudio con una resonancia magnética cardiaca con gadolinio, no siendo habitualmente necesario para el diagnóstico la realización de una biopsia cardiaca(5). El tratamiento de la amiloidosis cardiaca de elección es el trasplante cardiaco seguido de quimioterapia y trasplante autólogo de células hematopoyéticas, aunque la mayor parte de los pacientes no son candidatos a trasplante bien por la afectación multiorgánica de la enfermedad o bien por su edad. Ante esto, en función de la situación clínica del paciente, se debe valorar el inicio de quimioterapia con fines paliativos, y optimizar el tratamiento de la insuficiencia cardiaca para mejorar en la medida de lo posible el grado funcional del paciente(5-8).
Como vemos la afectación cardiaca en la amiloidosis AL confiere un marcado empeoramiento pronóstico, lo que justifica la necesidad de realizar un diagnóstico precoz para plantear una rápida terapéutica intensiva y global.
REFERENCIAS
- Rajkumar SV. Epidemiology, pathogenesis, clinical manifestations and diagnosis of Waldenström macroglobulinemia. [Monografía en Internet]. Walthman (MA): UpToDate 2014 Diciembre [acceso 15 de enero de 2015]. Disponible en: http://www.uptodate.com/
- Bladé Creixentí J, San Miguel JF. Gammapatías monoclonales. En: Farreras Valentí P, Rozman C, Editores. Medicina Interna. 17 ed. Barcelona: Elsevier; 2012. P.1626.
- Gertz MA, Kyle RA, Noel P. Primary systemic amyloidosis: a rare complication of immunoglobulin M monoclonal gammopathies and Waldenström’s macroglobulinemia. J Clin Oncol 1993;11(5):914-20.
- Rajkumar SV. Clinical presentation, laboratory manifestations, and diagnosis of immunoglobulin light chain (AL) amyloidosis (primary amyloidosis). [Monografía en Internet]. Walthman (MA): UpToDate 2014 Diciembre [acceso 15 de enero de 2015]. Disponible en: http://www.uptodate.com/
- Dubrey SW, Hawkins PN, Falk RH. Amyloid diseases of the heart: assessment, diagnosis, and referral. Heart 2011; 97(1):75-84.
- Wechalekar AD, Lachmann HJ, Goodman HJ, Bradwell A, Hawkins PN, Gillmore JD. AL amyloidosis associated with IgM paraproteinemia: clinical profile and treatment outcome. Blood 2008;112(10):4009-16.
- Dubrey SW, Cha K, Anderson J, Chamarthi B, Reisinger J, Skinner M, et al. The clinical features of immunoglobulin light-chain (AL) amyloidosis with heart involvement. QJM 1998;91(2):141-57.
- Falk RH. Clinical manifestations and diagnosis of amyloid cardiomyopathy. [Monografía en Internet]. Walthman (MA): UpToDate 2014 Diciembre [acceso 15 de enero de 2015]. Disponible en: http://www.uptodate.com/
INFORMACIÓN DEL ARTÍCULO
Autora para la correspondencia: Isabel Sánchez Berná. Servicio de Medicina Interna. Avenida de las Fuerzas Armadas, nº 2, CP 18014, Granada. E-mail: isabelsanchezberna@gmail.com





