

Revisión
Terapia génica suicida en la lucha contra el cáncer
Suicide gene therapy against cancer
Actual Med. 2021; 106(812): 54-65 DOI: 10.15568/am.2021.812.rev01
Recibido: 19/07/2020
Revisado: 23/07/2020
Aceptado: 14/09/2020
Resumen
El cáncer es una de las enfermedades con mayor mortalidad en todo el mundo, pero las terapias convencionales como la quimioterapia y la radioterapia no son eficaces en todos los casos y tienen amplios efectos secundarios. Para solventarlo se puede utilizar la terapia génica suicida, que consiste en inducir la muerte celular de las células cancerígenas mediante la introducción de un gen. Existen tres aproximaciones para el uso de esta terapia: la introducción de un gen codificante de un enzima generalmente bacteriano que activa un pro-fármaco, la introducción de un gen codificante de una toxina y la introducción de un gen pro-apoptótico. La expresión del gen se produce solamente en las células tumorales debido al uso de promotores tumor-específicos y vectores dirigidos. En estas tres aproximaciones se han desarrollado numerosos estudios con diversos sistemas llegando incluso a ensayos clínicos y productos aprobados para su uso en China (gendicina), viendo apoptosis de las células tumorales tanto in vitro como in vivo. Además, se han realizado mejoras en las técnicas mediante mutación de las enzimas y toxinas, modificación de los pro-fármacos y búsqueda de nuevas enzimas, toxinas o genes más activos, entre otros. A pesar de esto, se necesita seguir investigando en el campo para que la terapia sea totalmente dirigida y eficaz.
Palabras clave: Pro-fármaco; Apoptosis; Toxina.
Abstract
Cancers are a large family of diseases with the highest mortality rate worldwide. Conventional therapies such as chemotherapy or radiotherapy are not efficient in all cases and have important side effects. To solve it, suicide gene therapy can be used. This therapy consists on inducing cell death of cancer cells due to the introduction of a gene. There are three types of this therapy: introduction of a gene encoding generally a bacterial enzyme that actives a prodrug, introduction of a gene encoding a toxin or introduction of a proapoptotic gene. The expression of the targeted gene in tumor cells is produced by using tumor-specific promoters and target vectors. Using those three gene suicide therapies many hallmarks in the field were reached, achieving successful clinical trials and products approved to be used in China (gendicine), achieving apoptosis of tumor cells in vitro and in vivo. Furthermore, several improvements on these techniques were developed due to the mutation of the enzymes and toxins, modification of prodrugs and search of new more active enzymes, toxins and genes, between others. Regardless, further research on this area is needed to guarantee the efficiency of this state-of-the-art therapy and its effectiveness.
Keywords: prodrug; Apoptosis; Toxin.
Leer Artículo Completo
INTRODUCCIÓN
El cáncer es una de las enfermedades con más tasa de mortalidad, con más de 18 millones de casos nuevos y 9,6 millones de muertes en el año 2018 en todo el mundo (1). En España, el cáncer es la segunda causa de muerte (26,4 % de los fallecimientos), falleciendo más de 112.000 personas en el 2018 debido a un tumor. El número absoluto de diagnósticos de cáncer ha aumentado durante los años debido al aumento poblacional, el envejecimiento o la exposición a factores de riesgo (1).
Los tratamientos del cáncer más comunes implican cirugía, radioterapia y quimioterapia. La heterogeneidad de los tumores y la imposibilidad en muchos casos de un diagnóstico temprano son las mayores causas de que los tratamientos convencionales no tengan éxito. Además, suelen causar efectos secundarios. Por esto, es necesario desarrollar terapias más eficaces para combatir el cáncer (2).
La terapia génica se empezó a utilizar en los años 90 para tratar el cáncer para evitar estos problemas (3). El conjunto de técnicas se basa en introducir material genético en las células que componen el tumor para reprimir o activar la expresión de distintos genes diana para tratar la enfermedad. Existen numerosas estrategias para el tratamiento mediante terapia génica como la estimulación del sistema inmune, terapia génica antiangiogénica, corrección de defectos genéticos, inhibición de la invasión, entre otras (2).
Entre estas técnicas se encuentra la terapia génica suicida. Mediante esta metodología se introduce un gen que induce la apoptosis de la célula cancerígena de forma directa (introduciendo un gen que codifica una toxina o un gen pro-apoptótico) o de forma indirecta (necesitando un pro-fármaco, sustancia que se administra de forma sistémica y se activa en el tumor ejerciendo allí sus efectos) (2).
El objetivo de la presente revisión es analizar los diferentes sistemas utilizados para terapia génica suicida, así como las modificaciones que se han realizado para aumentar la eficacia de los mismos.
CUERPO DE LA REVISIÓN
Introducción de un gen activador de un pro-fármaco.
Mediante la estrategia denominada gene-directed enzyme prodrug therapy (GDEPT) las células tumorales son transfectadas con un gen que codifica para un enzima que no se sintetiza de forma natural en el resto de células sanas. Posteriormente, se administra un pro-fármaco, idealmente inocuo para el paciente, que es transformado por esta enzima solamente en las células transfectadas a un compuesto activo que induce la apoptosis de estas células (2, 4).
Para que la eficacia sea máxima el sistema enzima/pro-fármaco tiene que cumplir una serie de características:
- Especificidad. El pro-fármaco solamente debe ser modificada en el sitio del tumor (5).
- Utilizar un pro-fármaco no tóxico (5).
- Generar efecto espectador (5). Este efecto consiste en que la enzima producida por la célula afecta la célula transfectada y las adyacentes y es necesario debido a la baja eficacia de transfección, no llegando nunca al 100 %. Debido a este fenómeno, se ha observado una completa regresión del tumor después de la administración del pro-fárcaco incluso en casos en los que solo se han transfectado menos del 10 % de las células (2, 6). Se han observado diferentes mecanismos por los cuales se produce este efecto espectador: de forma directa, mediante procesos de difusión (7), transferencia por uniones tipo GAP (7, 8) y fagocitosis de vesículas apoptóticas (7) y de forma indirecta, por estimulación de la respuesta inmune (8).
- El compuesto activo debe atacar a las células proliferativas y a las no proliferativas para mantener la efectividad frente a tumores con crecimiento lento (5).
Ejemplos de sistemas enzima/pro-fármaco (tabla 1) son el linamarasa/linamarina (9), , el citocromo p450-4B1 con distintos pro-fármacos como el 4-ipomeanol (10) o el peroxidasa de rábano/indol-3-ácido acético (11), entre otros. Se describirán a continuación los sistemas enzima/ pro-fármacos más utilizados y estudiados actualmente.
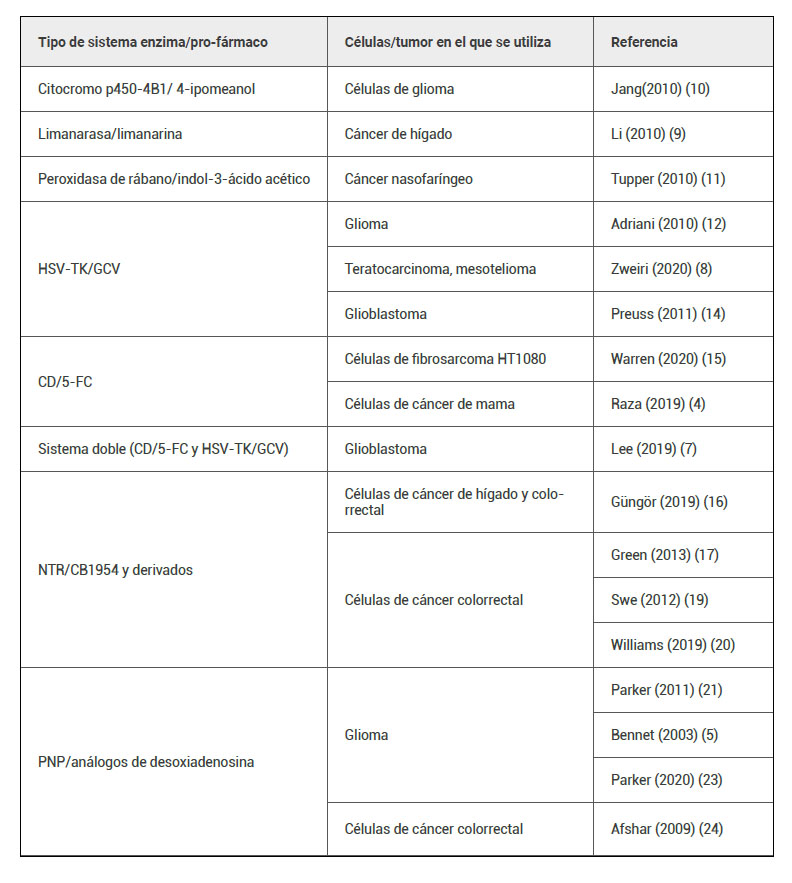
Tabla 1. Ejemplos de sistemas enzima/pro-fármaco y tipo de cáncer en el que se estudiaron.
Sistema timidina quinasa de virus del herpes simple y ganciclovir
Este sistema es uno de los más estudiados y utilizados, habiéndose realizado más de 70 ensayos clínicos, varios de ellos en fase III (12). Este sistema está formado por el gen codificante de la enzima timidina quinasa del virus del herpes simple (HSV-TK) y por el pro-fármaco ganciclovir (GCV). La HSV-TK convierte el pro-fármaco en GCV monofosfato y las quinasas celulares lo transforman en GCV trifosfato (figura 1).
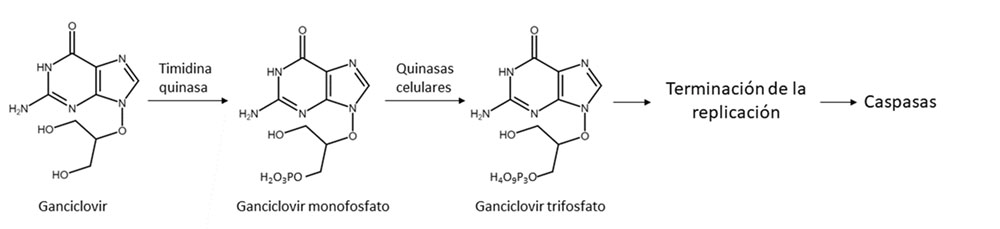
Figura 1. Esquema del funcionamiento del sistema HSV-TK/GCV.
El GCV trifosfato es utilizado por las ADN polimerasas α, δ y ϵ e incorporado al ADN, causando así la terminación de la replicación por la ausencia de desoxirribosa y, por lo tanto, ausencia de un extremo 3’-OH (13).
Esto promueve la apoptosis por la activación de las caspasas (6, 8).
Este sistema produce además efecto espectador mediante dos mecanismos distintos. El primero es un efecto directo a través de las uniones tipo GAP y de vesículas apoptóticas. El segundo mecanismo es un efecto indirecto debido a la estimulación del sistema inmune gracias a la presentación de antígenos específicos del tumor resultante de la muerte de las células HSV-TK positivas (8, 12).
A pesar de su amplio uso, este sistema tiene algunas limitaciones como la toxicidad inespecífica del transgén y del pro-fármaco, una cinética lenta, la muerte incompleta de todo el tumor y un efecto espectador más leve que el de otros sistemas (14). Además, la activación del pro-fármaco no es efectiva debido a la poca eficiencia catalítica del enzima utilizando GCV (12).
Para superar estas limitaciones se han desarrollado otras aproximaciones, como modificar la HSV-TK para mejorar la eficacia del sistema. Por ejemplo, tras inducir mutaciones en la secuencia de ADN se lograron sinterizar proteínas con mejores efectos terapéuticos tanto en experimentos in vitro como in vivo. Un ejemplo característico es la proteína TK.007, que requiere menos concentración de pro-fármaco y causa un mayor efecto espectador (14).
La enzima que cataliza la reacción de transformación del GCV monofosfato a GCV trifosfato es la enzima guanilato ciclasa. Por ello, se propuso la utilización de una proteína fusión expresando guanilato ciclasa y timidina quinasa (natural o modificada). Se observó que este sistema necesita una menor concentración de GCV y produce un aumento en el efecto espectador, ya que convierte más pro-fármaco en forma activa y se promueve la liberación de vesículas apoptóticas (12).
Sistema citosina desaminasa de Escherichia coli y 5-fluorocitosina
Este sistema se compone de la enzima citosina desaminasa (CD) de Escherichia coli y el pro-fármaco 5-fluorocitosina (5-FC), utilizada como antifúngico. La CD desamina la 5-FC, convirtiéndola en 5-fluorouracilo (5-FU). El 5-FU inhibe la timidilato sintasa, lo que conlleva una disminución de la concentración de desoxitimidina difosfato, roturas de doble cadena en el ADN y muerte celular (figura 2) (4, 6, 7).
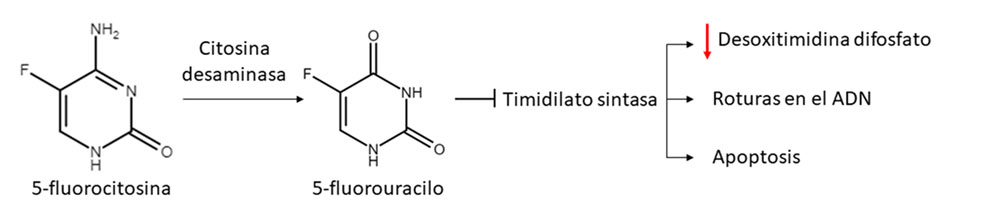
Figura 2. Esquema del funcionamiento del sistema CD/5-FC.
Este sistema produce un potente efecto espectador, ya que en un organismo modelo, el ratón, ha podido observarse la regresión completa de un tumor en el que solamente el 4 % de las células habían sido efectivamente transfectadas. Esto se puede deber a la difusión que 5-FU a través de la membrana plasmática y por tanto su efecto no es dependiente de uniones tipo GAP (4, 6).
Además, tiene ventajas sobre otros sistemas ya que el 5-FU actúa tanto como agente citotóxico como como radiosensibilizador, es decir, hace que las células tumorales sean más sensibles a la radioterapia (4).
Una de las desventajas de la CD es la baja afinidad por la 5-FC en comparación con la afinidad por la citosina, su sustrato natural. Por esto, se han identificado distintas enzimas mutantes que tienen menor afinidad por la citosina y más afinidad por la 5-FC, como la D314A o la F186W (4, 6). Para ir más allá, Raza y Ghosh mejoraron la actividad del sistema con la CD mutante F186W mediante la co-expresión del gen del enzima con el gen de la conexina-43. Esta conexina tiene un papel dual ya que promueve el efecto espectador debido a que forma parte de las uniones tipo GAP y, además, regula genes pro-apoptóticos en las células tumorales. A pesar de que el efecto espectador de este sistema no es dependiente de uniones tipo GAP, se observó una mejora en la eficacia del mismo (4).
Adicionalmente, se puede utilizar la enzima CD procedente de levaduras como f Saccharomyces cerevisiae, y realizar modificaciones de la enzima para que tengan mejores propiedades cinéticas y aumenten la eficacia del enzima (15).
Otro abordaje es la utilización de un sistema doble, como el que utiliza el sistema CD/5-FC en combinación con el sistema HSV-TK/GCV. Esta terapia doble aumenta la eficacia terapéutica aumentando el efecto espectador (7).
Sistema nitrorreductasa y CB1954
Los enzimas nitrorreductasas (NTR) catalizan la reducción de compuestos nitrogenados utilizando NAD(P)H (16). Cuando la 5-aziridinil-2,4-dinitrobenzamida (CB1954) se reduce por la acción de la NTR de E. coli, la más utilizada, se forman a partes iguales 2-hidroxilamina y 4-hidroxilamina (figura 3) (16). Estos dos metabolitos forman aductos en el ADN e inducen la apoptosis tanto en células tumorales quiescentes como replicantes (17).
La eficacia de estas enzimas ha sido probada en estudios clínicos debido a sus ventajas como por ejemplo, un fuerte efecto espectador. Sin embargo, este sistema presenta varios inconvenientes: el compuesto CB1954 es hepatotóxico y la eficacia de la NTR de E. coli es bastante baja. Así, se necesitan nuevos pro-fármacos alternativos o nuevas enzimas para aumentar la eficacia del sistema (16, 17).
Se ha visto que la nitrorreductasa MsuE de Pseudomonas aeruginosa, la NfsA de Neisseria meningitidis o la flavina reductasa FRasa I de Vibrio fischeri son más efectivas que la de E. coli (17,18,19).
Además, también se han realizado estudios de mutagénesis para conseguir enzimas con mayor actividad, tanto con el enzima de E. coli como con otras reductasas como la FRasa I, necesitando una menor concentración de pro-fármaco para ser efectivas (19, 20).
Sistema purina nucleósido fosforilasa y 6-metilpurina desoxirribósido o fludarabina
La enzima purina nucleósido fosforilasa (PNP) de E. coli cataliza la conversión de análogos de desoxiadenosina que no son tóxicos en compuestos tóxicos para la célula. Los análogos más comunes son la 6-metilpurina-2’-desoxirribonucleósido (MeP-dR) y la fludarabina (6, 21). La PNP rompe el enlace glicosídico de los análogos liberando metilpurina o 2- fluoroadenina, dependiendo del pro-fármaco utilizado. En células humanas, estos compuestos se convierten en sus respectivos trifosfatos debido a la acción secuencial de varias enzimas celulares. Estos compuestos trifosforilados inhiben la síntesis de ARN y de proteínas, siendo eficaces frente a células que no están en proceso de división, actuando frente a las células madre tumorales que se encuentran mayoritariamente en estado de quiescencia (21).
Este sistema presenta un potente efecto espectador. Esto se debe a que tanto la metilpurina como la 2- fluoroadenina pueden difundir libremente a través de las membranas celulares, llegando así fácilmente a las células vecinas (21).
El empleo de este sistema llegó a la fase I de ensayos clínicos utilizando el pro-fármaco fludarabina, observando una regresión del tumor en los pacientes. La fludarabina tiene bastante interés como pro-fármaco ya que está aprobado para el tratamiento de leucemias linfocíticas crónicas (6, 21).
A pesar de esto, se necesita generar nuevos pro-fármacos que sean menos tóxicos y que tengan una circulación sistémica prolongada, además de producir PNPs modificadas para aumentar la eficacia (21). Por ello, se han creado PSPs mutantes como la M64V PNP, que utiliza un análogo de desoxiadenosina con 100 veces más eficiencia que la PNP normal (5). Para esta enzima se diseñaron y evaluaron más pro-fármacos que pudiesen aumentar la eficacia del sistema, aunque siguen siendo relativamente tóxicas (22).
Además, se utilizaron PNPs de otras especies como Trichomonas vaginalis, la cual presenta 25 veces más eficiencia catalítica utilizando la fludarabina que la PNP de E. coli, obteniendo una gran eficiencia de regresión tumoral in vivo (23). Incluso se ha utilizado una PNP humana mutada que reconoce derivados de desoxiadenosina y que podría producir menos reacción inmunológica que las enzimas bacterianas utilizadas (24).
Introducción de un gen codificante de una toxina
Esta aproximación se basa en la introducción de forma selectiva en las células cancerígenas de un gen que codifica una toxina, funcionando esta como agente quimioterapéutico. El producto solamente se genera en las células cancerígenas, dejando a las células normales intactas. Este sistema solo se puede utilizar cuando el gen de la toxina de interés está bien caracterizado. La ventaja de este sistema es que no necesita un tratamiento con un pro-fármaco, por lo que los problemas relativos al mismo así como, los efectos secundarios o la biodisponibilidad limitada se eliminan, aunque puede producir efectos secundarios como reacciones inflamatorias incontroladas (2).
Muchos microorganismos producen de manera natural toxinas que afectan a las células de mamífero, siendo así susceptibles de ser utilizadas en este tipo de terapia génica (2). Ejemplos de toxinas bacterianas utilizadas para este tipo de terapia (tabla 2) son la enterotoxina de Clostridium perfingens (25) o la estreptolisina O de Streptococcus sp (26). Incluso se utilizaron toxinas derivadas de virus, como el gen E del virus phiX174 (27) o la apoptina del virus de la anemia del pollo (28). Se describirán a continuación las toxinas más utilizadas.
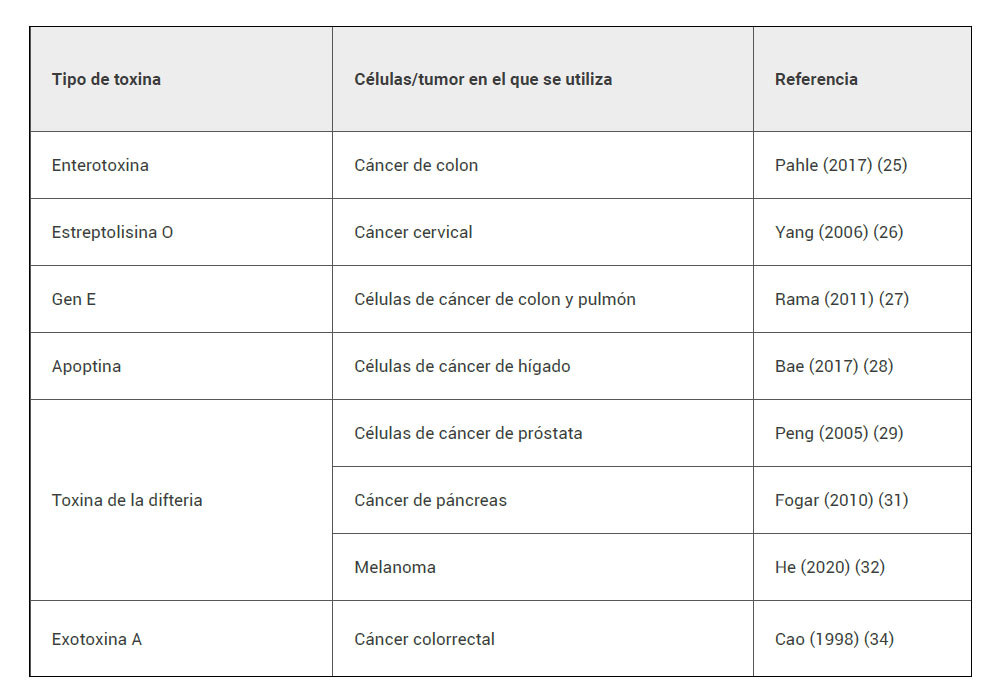
Tabla 2. Ejemplos de toxinas y tipo de cáncer en el que se estudiaron.
Toxina de la difteria
Es una de las más utilizadas en terapia génica suicida (29). Es producida y secretada de forma natural por Corynebacterium diphtheriae como un polipéptido precursor que se rompe enzimáticamente en dos cadenas: A y B. La cadena B se une a la superficie de la mayoría de células eucariotas, liberando la cadena A en el citoplasma. Una vez dentro de la célula, la cadena A inhibe la síntesis de proteínas ya que inactiva el factor de elongación 2 (EF2) mediante su ADP-ribosilación, lo que conlleva la muerte celular tanto en células en división como en células quiescentes (29, 30). En este tipo de terapia solamente se introduce la parte del gen codificante de la cadena A. En ausencia de la cadena B, la cadena A no puede ejercer su efecto en células vecinas así que la producción está limitada a las células diana (29).
Para que la toxina solamente induzca apoptosis en las células tumorales, se necesita un sistema de expresión específico. Peng y colaboradores (29) desarrollaron un sistema de recombinación y expresión bajo el control del promotor del gen codificante para el antígeno específico de próstata (PSA). Así, observaron expresión de la toxina solamente en células que expresaban este gen.
Para aplicar con éxito estas terapias es necesario que un método de transfección eficiente o combinarlo con otras terapias convencionales (29).
A pesar de que se han desarrollado diversos promotores específicos de tumores, en algunos casos es preferible controlar de manera exógena la expresión del gen codificante de la toxina. Por ejemplo, se ha utilizado el promotor de la familia de proteínas heat shock 70 (HSP70) en conjunto con el gen de la cadena A como posible tratamiento para el cáncer de páncreas. Este promotor es útil ya que HSP70 es expresada por las células del cáncer de páncreas, se puede activar la expresión por calor, es muy eficiente (impidiendo el crecimiento celular en el 100% de células transfectadas) y se puede controlar de forma espacial focalizando la fuente de calor en el sitio del tumor (31). Esta metodología ha sido usada con éxito para tratar casos de cáncer de páncreas consiguiéndose resultados prometedores in vivo (31).
Posteriormente, se ha utilizado un sistema de transgénesis que se regula con luz. Este sistema se compone de dos partes: un factor de transcripción sensible a la luz y un promotor específico que, al iluminarlos con luz azul, inicien la transcripción de los genes de interés. Este sistema tiene una gran eficiencia de inducción, una cinética rápida y no interfiere con la señalización celular. Puede ser muy útil sobre todo para cánceres como melanomas. Recientemente se ha utilizado este sistema introduciéndolo en el tumor mediante nanopartículas y se ha visto eficacia antitumoral in vivo, además de que la expresión de la toxina es dependiente de la intensidad de la irradiación (32).
Exotoxina A
La exotoxina A es sintetizada por P. aeruginosa. Se produce como una única molécula polipeptídica compuesta por tres dominios estructurales y funcionales (I, II y III), que participan en el proceso de intoxicación. Este proceso comienza con la unión del dominio I a la proteína relacionada con el receptor de la lipoproteína de baja densidad (LRP). Este receptor permite la endocitosis de la exotoxina A, lo que promueve un cambio de conformación debido al cambio de pH. Esto hace que el dominio II se inserte en la membrana del endosoma. Posteriormente pueden ocurrir dos procesos: la toxina puede cruzar la membrana del endosoma o puede ser procesada, generando dos fragmentos, uno de ellos conteniendo parte del dominio II y el dominio enzimático III.
Una vez en el citosol, el dominio catalítico inactiva mediante ADP-ribosilación al EF2, inactivando la síntesis de proteínas y produciendo la muerte celular (33).
La parte codificante de los dominios I y II de la exotoxina A fue expresada frente al promotor del antígeno carcinoembrionario humano (CEA) en células de carcinoma colorrectal, ya que es este tipo de tumores este antígeno está sobre-expresado, observandose una reducción del tamaño de los tumores en condiciones in vivo (34).
Introducción de un gen pro-apoptótico
En esta aproximación se introducen genes pro-apoptóticos o genes supresores de tumores de forma específica en las células tumorales, que sufren un déficit en la señalización de la apoptosis. Los genes supresores de tumores regulan negativamente oncogenes, deceleran la división celular, reparan errores en el ADN e inducen la muerte celular cuando es necesaria. Así, los productos de estos genes protegen a la célula de convertirse en una célula cancerígena (2).
Ejemplos de genes pro-apoptóticos utilizados en terapia génica suicida (tabla 3) son los codificantes de Smac (35), la caspasa 3 (36) o Bax (37), entre otros. Los genes pro-apoptóticos más utilizados son:
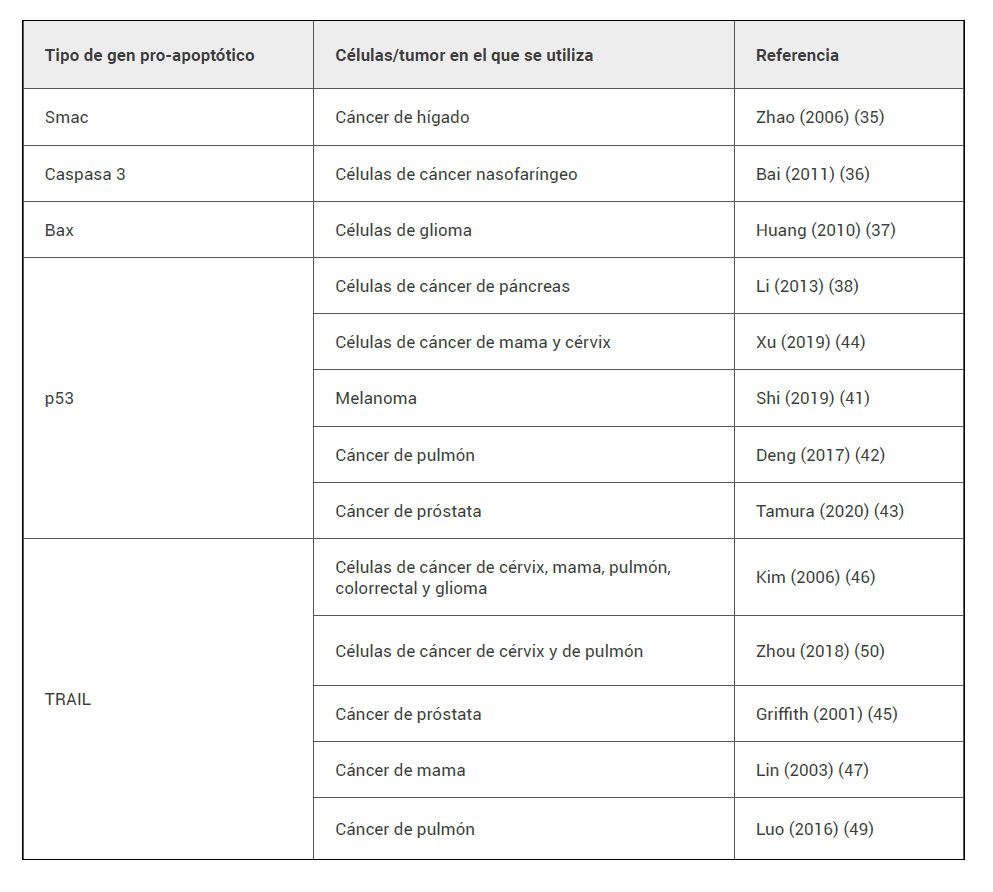
Tabla 3. Ejemplos de genes pro-apoptóticos y tipo de cáncer en el que se estudiaron.
p53
p53 es uno de los genes más utilizados en este tipo de terapias ya que se encuentra mutado en la mayoría de células cancerígenas (38, 39). Es denominado el “guardián del genoma”, ya que induce el arresto del ciclo celular, la senescencia y la apoptosis en respuesta a la activación de oncogenes, daño en el ADN y otras señales de estrés, como pueden ser la radio y la quimioterapia (39).
Las vías de muerte celular inducidas por la expresión de p53 están involucradas en la supresión de la iniciación y desarrollo del tumor. Un gran número de genes están identificados como genes diana de p53, algunos involucrados en la apoptosis (Bax o Noxa), en el arresto del ciclo celular (p21) o en la autofagia (DRAM) (39).
La gendicina, desarrollada por Shenzhen SiBiono GeneTech Co. Ltd., es una terapia que introduce el gen p53 natural humano mediante un adenovirus recombinante (Ad5RSV-p53) aprobada para su uso clínico en China en 2003 para el tratamiento de cáncer de cabeza y cuello. Más de 30.000 pacientes han sido tratados con este tipo de terapia, viendo respuesta en el 90-96 % de los casos (40). Además, se están llevando a cabo estudios clínicos con otro tipo de vectores y células diana (39). Por ejemplo, recientemente se realizaron estudios clínicos probando esta terapia frente a melanoma, viendo inhibición del crecimiento del tumor sin efectos secundarios (41).
Este tipo de terapia se está empleando en varios estudios para eliminar las células cancerígenas remanentes tras la extirpación quirúrgica de tumores de pulmón, ya que la recurrencia y la metástasis son las principales causas de muerte tras la cirugía. En este estudio se comprobó la eficacia del tratamiento adyuvante con p53, reduciendo significativamente la recurrencia y la metástasis (42).
La introducción de este gen favorece la radiosensibilización de células cancerígenas pancreáticas, favoreciendo su eliminación mediante radioterapia por un aumento en la expresión de p21 y Bax (38).
Esta terapia también se está probando con otro tipo de agentes quimioterapéuticos. Esta cooperación puede conllevar una mayor eficacia utilizando menores dosis de agente quimioterapéutico, minimizando así los efectos secundarios. Por ejemplo, se probó junto con el cabazitaxel, un fármaco utilizado frente a cáncer de páncreas. Esta combinación bloquea el progreso del tumor in vivo utilizando una dosis baja de cabazitaxel (12,5 mg/kg) (43). También se ha utilizado en combinación con el resveratrol, un compuesto natural caracterizado por la inhibición del crecimiento de células cancerígenas de cáncer de mama, colon, ovario y pulmón. La apoptosis inducida por el resveratrol está relacionada con la vía de p53. Esta combinación se ha utilizado in vitro frente a células cancerígenas de cérvix y de mama. Se observó un aumento en la inducción de la apoptosis e inhibición del crecimiento del tumor en comparación con cada terapia por separado (44).
TRAIL
Unos de los agentes inductores de apoptosis mejor caracterizados son los pertenecientes a la familia del factor de necrosis tumoral (TNF), como el ligando inductor de la apoptosis relacionado con TNF (TRAIL), codificado por el gen TNFSF10 (miembro 10 de la superfamilia del factor de necrosis tumoral). TNFSF10 es expresado en muchos tipos celulares y tejidos de manera constitutiva (45).
TRAIL forma un homotrímero, el cual activa la caspasa 8, la cual inicia cascadas de señalización que inducen la rotura de proteínas del citoplasma, del citoesqueleto y del núcleo, así como la rotura del ADN. Para esto, TRAIL tiene que formar un homotrímero. TRAIL tiene una gran selectividad para inducir la apoptosis solamente en células tumorales, tanto in vitro como in vivo, por lo que no se detecta citotoxicidad en modelos animales (46).
Se ha observado que esta técnica es eficaz in vivo. En varios modelos de cáncer de próstata se observó que esta terapia inducía la apoptosis de las células tumorales (45).
Como la formación del homotrímero es esencial para su función, se ha creado una construcción génica a partir de TNFSF10 con un dominio adicional de trimerización que hace más fácil la formación de estos complejos. Así, se ha observado que la inducción de la apoptosis era más eficaz. Utilizando un adenovirus como vector, se observó la supresión del crecimiento tumoral de glioma tanto in vitro como in vivo (46).
Además, se ha visto que la combinación de esta terapia génica con terapias tradicionales tiene un efecto sinérgico. Por ejemplo, frente al cáncer de mama y al cáncer de pulmón metastático derivado de cáncer de mama se observó un efecto adictivo in vivo en la utilización de esta terapia génica combinada con agentes quimioterapéuticos como la hidrocortisona (47).
Recientemente se han investigado otro tipo de vectores para mejorar la eficacia de la transfección. En un estudio se utilizó un lípido catiónico (1,2-di-(9Z-octadecenoil)-3-biguanida-propano) fusionado con metformina, fármaco que se utiliza para pacientes con diabetes tipo II, pero que tiene un efecto antitumoral (48). Así, este sistema presenta una mejor eficacia antitumoral que utilizando como vector lípidos catiónicos convencionales tanto in vitro como in vivo (49).
También se han utilizado polímeros funcionales que responden a estímulos intracelulares y liberan más fácilmente el ADN como el polímero B-PDEAEA, que responde a los niveles altos de especies reactivas de oxígeno (ROS) presentes en las células tumorales. Para aumentar la producción de ROS por las células tumorales y que la liberación del ADN sea más eficiente, se utilizó esta aproximación con el enzima SAHA, una histona deacetilasa que está aprobada para el tratamiento de algunos cánceres y que produce un aumento en la concentración de ROS de las células tumorales (50).
CONCLUSIONES
El cáncer es una de las enfermedades que presenta una mayor tasa de mortalidad en todo el mundo, presentando las actuales terapias convencionales una efectividad limitada. La terapia génica suicida es una técnica que tiene mucho potencial en el tratamiento de esta enfermedad gracias a sus múltiples aproximaciones de las cuales se están realizando diferentes ensayos clínicos. Los tres tipos de terapia génica suicida cuentan con resultados prometedores, aunque la introducción de un gen proapoptótico (en concreto p53) parece la más adelantada ya que se ha aprobado para su uso clínico en China.
Aun así, este tipo de terapia todavía no es del todo eficiente, por lo que hay que seguir investigando y realizando ensayos clínicos sobre los sistemas actuales y desarrollando nuevos métodos para que esta terapia sea completamente segura y eficaz.
REFERENCIAS BIBLIOGRÁFICAS
- ↑ SEOM. Las cifras del cáncer en España 2020. Madrid: Sociedad Española de Oncología Médica; 2020. p. 35.
- ↑ Gholami A. Suicide gene therapy: A special focus on progress and concerns about cancer treatment. Trends Pharm Sci. 2017;3(4):221-36.
- ↑ Rosenberg SA, Aebersold P, Cornetta K, et al. Gene transfer into humans–immunotherapy of patients with advanced melanoma, using tumor-infiltrating lymphocytes modified by retroviral gene transduction. N Engl J Med. 1990;323(9):570-8. DOI: 10.1056/NEJM199008303230904
- ↑ Raza A, Ghosh SS. Connexin-43 Enhances the Redesigned Cytosine Deaminase Activity for Suicide Gene Therapy in Human Breast Cancer Cells. Biochem Insights. 2019;12:1-8. DOI: 10.1177/1178626418818182
- ↑ Bennett EM, Anand R, Allan PW, et al. Designer gene therapy using an Escherichia coli purine nucleoside phosphorylase/prodrug system. Chem Biol. 2003;10(12):1173-81. DOI: 10.1016/j.chembiol.2003.11.008
- ↑ Duzgunes N. Suicide Gene Therapy: Methods and Protocols. 1 ed. New York: Humana Press; 2019. 190 p.
- ↑ Lee M, Kim Y-S, Lee K, et al. Novel Semi-Replicative Retroviral Vector Mediated Double Suicide Gene Transfer Enhances Antitumor Effects in Patient-Derived Glioblastoma Models. Cancers. 2019;11(8):1090. DOI: 10.3390/cancers11081090
- ↑ Zweiri J, Christmas SE. Demonstration of anti-tumour bystander killing with prodrug-preloaded suicide gene-engineered tumour cells: a potential improvement for cancer therapeutics. Cancer cell international. 2020;20:26-. DOI: 10.1186/s12935-020-1115-4
- ↑Li J, Li H, Zhu L, et al. The adenovirus-mediated linamarase/linamarin suicide system: A potential strategy for the treatment of hepatocellular carcinoma. Cancer Lett. 2010;289(2):217-27. DOI: 10.1016/j.canlet.2009.08.016
- ↑ Jang SJ, Kang JH, Lee TS, et al. Prodrug-activating Gene Therapy with Rabbit Cytochrome P450 4B1/4-Ipomeanol or 2-Aminoanthracene System in Glioma Cells. Nucl Med Mol Imaging. 2010;44(3):193-8. DOI: 10.1007/s13139-010-0038-8
- ↑ Tupper J, Stratford MR, Hill S, Tozer GM, Dachs GU. In vivo characterization of horseradish peroxidase with indole-3-acetic acid and 5-bromoindole-3-acetic acid for gene therapy of cancer. Cancer Gene Ther. 2010;17(6):420-8. DOI: 10.1038/cgt.2009.86
- ↑ Ardiani A, Sanchez-Bonilla M, Black ME. Fusion enzymes containing HSV-1 thymidine kinase mutants and guanylate kinase enhance prodrug sensitivity in vitro and in vivo. Cancer Gene Ther. 2010;17(2):86-96. DOI: 10.1038/cgt.2009.60
- ↑ Ilsley DD, Lee S-H, Miller WH, Kuchta RD. Acyclic Guanosine Analogs Inhibit DNA Polymerases .alpha., .delta., and .epsilon. with Very Different Potencies and Have Unique Mechanisms of Action. Biochemistry. 1995;34(8):2504-10. DOI: 10.1021/bi00008a014
- ↑ Preuss E, Muik A, Weber K, Otte J, von Laer D, Fehse B. Cancer suicide gene therapy with TK.007: superior killing efficiency and bystander effect. J Mol Med (Berl). 2011;89(11):1113-24. DOI: 10.1007/s00109-011-0777-8
- ↑ Warren TD, Patel K, Rivera JL, Eshleman JR, Ostermeier M. Comprehensive mutagenesis on yeast cytosine deaminase yields improvements in 5-fluorocytosine toxicity in HT1080 cells. AIChE Journal. 2020;66(3):e16688. DOI: 10.1002/aic.16688
- ↑ Güngör T, Önder FC, Tokay E, et al. Prodrugs for nitroreductase based cancer therapy- 2: Novel amide/Ntr combinations targeting PC3 cancer cells. Eur J Med Chem. 2019;171:383-400. DOI: 10.1016/j.ejmech.2019.03.035
- ↑ Green LK, Storey MA, Williams EM, et al. The Flavin Reductase MsuE Is a Novel Nitroreductase that Can Efficiently Activate Two Promising Next-Generation Prodrugs for Gene-Directed Enzyme Prodrug Therapy. Cancers. 2013;5(3):985-97. DOI: 10.3390/cancers5030985
- ↑ Rich MH, Sharrock AV, Hall KR, Ackerley DF, MacKichan JK. Evaluation of NfsA-like nitroreductases from Neisseria meningitidis and Bartonella henselae for enzyme-prodrug therapy, targeted cellular ablation, and dinitrotoluene bioremediation. Biotechnol Lett. 2018;40(2):359-67. DOI: 10.1007/s10529-017-2472-5
- ↑ Swe PM, Copp JN, Green LK, et al. Targeted mutagenesis of the Vibrio fischeri flavin reductase FRase I to improve activation of the anticancer prodrug CB1954. Biochem Pharmacol. 2012;84(6):775-83. DOI: 10.1016/j.bcp.2012.07.002
- ↑ Williams EM, Rich MH, Mowday AM, et al. Engineering Escherichia coli NfsB To Activate a Hypoxia-Resistant Analogue of the PET Probe EF5 To Enable Non-Invasive Imaging during Enzyme Prodrug Therapy. Biochemistry. 2019;58(35):3700-10. DOI: 10.1021/acs.biochem.9b00376
- ↑ Parker WB, Allan PW, Waud WR, Hong JS, Sorscher EJ. Effect of expression of adenine phosphoribosyltransferase on the in vivo anti-tumor activity of prodrugs activated by E. coli purine nucleoside phosphorylase. Cancer Gene Ther. 2011;18(6):390-8. DOI: 10.1038/cgt.2011.4
- ↑ Parker WB, Allan PW, Ealick SE, et al. Design and evaluation of 5’-modified nucleoside analogs as prodrugs for an E. coli purine nucleoside phosphorylase mutant. Nucleosides Nucleotides Nucleic Acids. 2005;24(5-7):387-92. DOI: 10.1081/ncn-200059807
- ↑ Parker WB, Allan PW, Waud WR, et al. The use of Trichomonas vaginalis purine nucleoside phosphorylase to activate fludarabine in the treatment of solid tumors. Cancer Chemother Pharmacol. 2020;85(3):573-83. DOI: 10.1007/s00280-019-04018-7
- ↑ Afshar S, Asai T, Morrison SL. Humanized ADEPT comprised of an engineered human purine nucleoside phosphorylase and a tumor targeting peptide for treatment of cancer. Molecular cancer therapeutics. 2009;8(1):185-93. DOI: 10.1158/1535-7163.MCT-08-0652
- ↑ Pahle J, Menzel L, Niesler N, et al. Rapid eradication of colon carcinoma by Clostridium perfringens Enterotoxin suicidal gene therapy. BMC Cancer. 2017;17(1):129. DOI: 10.1186/s12885-017-3123-x
- ↑ Yang WS, Park SO, Yoon AR, et al. Suicide cancer gene therapy using pore-forming toxin, streptolysin O. Molecular cancer therapeutics. 2006;5(6):1610-9. DOI: 10.1158/1535-7163.MCT-05-0515
- ↑ Rama AR, Prados J, Melguizo C, et al. E phage gene transfection associated to chemotherapeutic agents increases apoptosis in lung and colon cancer cells. Bioeng Bugs. 2011;2(3):163-7. DOI: 10.4161/bbug.2.3.15533
- ↑ Bae Y, Song SJ, Mun JY, Ko KS, Han J, Choi JS. Apoptin Gene Delivery by the Functionalized Polyamidoamine (PAMAM) Dendrimer Modified with Ornithine Induces Cell Death of HepG2 Cells. Polymers. 2017;9(6):197. DOI: 10.3390/polym9060197
- ↑ Peng W, Chen J, Huang YH, Sawicki JA. Tightly-regulated suicide gene expression kills PSA-expressing prostate tumor cells. Gene Ther. 2005;12(21):1573-80. DOI: 10.1038/sj.gt.3302580
- ↑ Pappenheimer AM, Jr. Diphtheria toxin. Annu Rev Biochem. 1977;46:69-94. DOI: 10.1146/annurev.bi.46.070177.000441
- ↑ Fogar P, Navaglia F, Basso D, et al. Heat-induced transcription of diphtheria toxin A or its variants, CRM176 and CRM197: implications for pancreatic cancer gene therapy. Cancer Gene Ther. 2010;17(1):58-68. DOI: 10.1038/cgt.2009.48
- ↑ He M, Wang Y, Chen X, et al. Spatiotemporally controllable diphtheria toxin expression using a light-switchable transgene system combining multifunctional nanoparticle delivery system for targeted melanoma therapy. J Control Release. 2020;319:1-14. DOI: 10.1016/j.jconrel.2019.12.015
- ↑ Morlon-Guyot J, Méré J, Bonhoure A, Beaumelle B. Processing of Pseudomonas aeruginosa exotoxin A is dispensable for cell intoxication. Infection and immunity. 2009;77(7):3090-9. DOI: 10.1128/IAI.01390-08
- ↑ Cao GW, Qi ZT, Pan X, et al. Gene therapy for human colorectal carcinoma using human CEA promoter contro led bacterial ADP-ribosylating toxin genes human CEA: PEA & DTA gene transfer. World J Gastroenterol. 1998;4(5):388-91. DOI: 10.3748/wjg.v4.i5.388
- ↑ Zhao J, Jin J, Zhang X, et al. Transfection of Smac sensitizes tumor cells to etoposide-induced apoptosis and eradicates established human hepatoma in vivo. Cancer Gene Ther. 2006;13(4):420-7. DOI: 10.1038/sj.cgt.7700910
- ↑ Bai D, Xia X, Yow CMN, Chu ESM, Xu C. Hypocrellin B-encapsulated nanoparticle-mediated rev-caspase-3 gene transfection and photodynamic therapy on tumor cells. Eur J Pharmacol. 2011;650(2):496-500. DOI: 10.1016/j.ejphar.2010.10.017
- ↑ Huang J, Gao J, Lv X, et al. Target gene therapy of glioma: overexpression of BAX gene under the control of both tissue-specific promoter and hypoxia-inducible element. Acta Biochim Biophys Sin. 2010;42(4):274-80. DOI: 10.1093/abbs/gmq016
- ↑ Li J, Pan J, Zhu X, et al. Recombinant adenovirus-p53 (Gendicine) sensitizes a pancreatic carcinoma cell line to radiation. Chinese journal of cancer research. 2013;25(6):715-21. DOI: 10.3978/j.issn.1000-9604.2013.11.12
- ↑ Chen GX, Zhang S, He XH, Liu SY, Ma C, Zou XP. Clinical utility of recombinant adenoviral human p53 gene therapy: current perspectives. OncoTargets Ther. 2014;7:1901-9. DOI: 10.2147/OTT.S50483
- ↑ Zhang W-W, Li L, Li D, et al. The First Approved Gene Therapy Product for Cancer Ad-p53 (Gendicine): 12 Years in the Clinic. Hum Gene Ther. 2018;29(2):160-79. DOI: 10.1089/hum.2017.218
- ↑ Shi SW, Li B, Dong Y, et al. In Vitro and Clinical Studies of Gene Therapy with Recombinant Human Adenovirus-p53 Injection for Malignant Melanoma. Hum Gene Ther Clin Dev. 2019;30(1):7-18. DOI: 10.1089/humc.2018.112
- ↑Deng B, Sun T, Tang B, et al. Surgery combined with adenoviral p53 gene therapy for treatment of non-small cell lung cancer: a phase II study. Oncotarget. 2017;8(63):107089-95. DOI: 10.18632/oncotarget.22333
- ↑ Tamura RE, Lana MG, Costanzi-Strauss E, Strauss BE. Combination of cabazitaxel and p53 gene therapy abolishes prostate carcinoma tumor growth. Gene Ther. 2020;27(1):15-26. DOI: 10.1038/s41434-019-0071-x
- ↑ Xu X, Liu A, Bai Y, et al. Co-delivery of resveratrol and p53 gene via peptide cationic liposomal nanocarrier for the synergistic treatment of cervical cancer and breast cancer cells. J Drug Deliv Sci Technol. 2019;51:746-53. DOI: 10.1016/j.jddst.2018.05.008
- ↑ Griffith TS, Broghammer EL. Suppression of Tumor Growth Following Intralesional Therapy with TRAIL Recombinant Adenovirus. Mol Ther. 2001;4(3):257-66. DOI: 10.1006/mthe.2001.0439
- ↑ Kim CY, Jeong M, Mushiake H, et al. Cancer gene therapy using a novel secretable trimeric TRAIL. Gene Ther. 2006;13(4):330-8. DOI: 10.1038/sj.gt.3302658
- ↑ Lin T, Zhang L, Davis J, et al. Combination of TRAIL gene therapy and chemotherapy enhances antitumor and antimetastasis effects in chemosensitive and chemoresistant breast cancers. Mol Ther. 2003;8(3):441-8. DOI: 10.1016/s1525-0016(03)00203-x
- ↑ Morales DR, Morris AD. Metformin in Cancer Treatment and Prevention. Annu Rev Med. 2015;66(1):17-29. DOI: 10.1146/annurev-med-062613-093128
- ↑ Luo C, Miao L, Zhao Y, et al. A novel cationic lipid with intrinsic antitumor activity to facilitate gene therapy of TRAIL DNA. Biomaterials. 2016;102:239-48. DOI: 10.1016/j.biomaterials.2016.06.030
- ↑ Zhou X, Liu Z, Wang H, et al. SAHA (vorinostat) facilitates functional polymer-based gene transfection via upregulation of ROS and synergizes with TRAIL gene delivery for cancer therapy. J Drug Target. 2018;27(3):306-14. DOI: 10.1080/1061186X.2018.1519028
INFORMACIÓN DEL ARTÍCULO
Conflicto de intereses: El autor/a de este artículo declara no tener ningún tipo de conflicto de intereses respecto a lo expuesto en el presente trabajo.
Autora para la correspondencia: Celia Piñeiro Silva. Departamento de Fisiología. Facultad de Veterinaria. Universidad de Murcia. Campus de Espinardo · 30100, Murcia, España. E-mail: celia.pineiros@um.es





