

Revisión
Mecanismos de inactivación del cromosoma X. Implicaciones y consecuencias clínicas
X Chromosome Inactivation mechanisms. Clinical implications and consequences
Actual Med.2025;110(821):22-30 DOI: 10.15568/am.2025.821.rev02
Recibido: 15/07/2024
Revisado: 21/02/2025
Aceptado: 09/03/2025
Resumen
La inactivación del cromosoma X (ICX) es el proceso que permite la compensación de dosis génica entre machos y hembras en numerosas especies y uno de los mecanismos genéticos fundamentales para la vida. Actualmente, el papel de la genética en la práctica clínica está creciendo exponencialmente, estando el proceso de ICX cada vez más presente en las investigaciones en este ámbito.
Los recientes estudios sobre la inactivación del cromosoma X en humanos relacionan este proceso con mucha más patología de la que se pensaba inicialmente. Algunos ejemplos de esta patología como el cáncer, las enfermedades ligadas al cromosoma X o las cada vez más diagnosticadas enfermedades autoinmunes son solo algunas de las líneas de investigación que acercan el proceso de ICX a la práctica clínica.
En esta revisión se intenta resumir los principales mecanismos que rigen el proceso de ICX, además de revisar qué relevancia tiene este proceso en ciertas patologías y sintetizar algunas de las posibles líneas de investigación futuras sobre este tema.
Palabras clave: Inactivación del cromosoma X; XIST; Enfermedades ligadas al X; Desarrollo embrionario.
ABSTRACT
X-chromosome inactivation (XCI) is the process that allows gene dose compensation between males and females in numerous species and one of the basic genetic mechanisms for life. Currently, the role of genetics in clinical practice is growing exponentially, with the ICX process being increasingly present in research in this field.
Recent studies on X-chromosome inactivation in humans relate this process to much more pathology than it was initially thought. Some examples of this pathology such as cancer, X-linked diseases or the increasingly diagnosed autoimmune diseases are just some of the lines of research that bring the ICX process closer to clinical practice.
This paper attempts to summarize the main mechanisms that govern the ICX process, as well as to review the relevance of this process in certain diseases and to synthesize some of the possible future research lines on this topic.
Keywords: X-chromosome inactivation; XIST non-coding RNA; Genetic diseases X-linked; Embryonic development.
Leer Artículo Completo
INTRODUCCIÓN
La inactivación del cromosoma X (ICX) o Lyonización, es un proceso que ocurre durante el desarrollo embrionario en las hembras para compensar el desequilibrio genético entre sexos (1). Aunque frecuentemente se usa este término “inactivación del cromosoma X o “X inactivation” en inglés para referirse a este fenómeno, Barbara R. Migeon et al (2) diferencian esta expresión de otra: “single active X”, la cual se ajusta más a cómo ocurre este proceso en nuestra especie. Según los autores, los mecanismos que participan en este proceso no eligen qué cromosoma X inactivar, sino más bien cuál de ellos se mantendrá activo.
Este fenómeno fue descrito por primera vez por Mary Lyon, quien postuló que solo un cromosoma X de cada célula se inactiva aleatoriamente, pudiendo ser materno o paterno, resultando las mujeres mosaicos para el cromosoma X (3). Un ejemplo de este mecanismo se encuentra en las gatas “calicó”, que presentan manchas naranjas y negras en la piel, correspondiendo cada color a un grupo celular: uno en la que está activo el cromosoma X con el alelo naranja y en otro grupo, el negro (3). Como este proceso ocurre solo en células con varios cromosomas X, no encontramos gatos macho (4).
Fue en esta misma especie en la que Murray Barr, en 1949, descubrió los corpúsculos de Barr. Se definen como un tipo de heterocromatina, sólo presente en células con dos cromosomas X, que correspondería al cromosoma que es inactivado para conseguir la compensación de la carga génica (5). Así, si una célula tiene un exceso de cromosomas X (por ejemplo, tres), se inactivan todos los sobrantes (en el ejemplo, dos).
Sin embargo, este hallazgo plantea otra cuestión: si se inactivan los cromosomas X sobrantes, ¿por qué personas con patologías en las que se encuentran un exceso de cromosomas sexuales femeninos están afectadas? (3). Esto se explica teniendo en cuenta que la inactivación del cromosoma X es incompleta, pues aproximadamente un 12% de los genes escapan de este proceso en humanos (6).
Todos estos hallazgos comentados fueron descubiertos gracias al estudio en animales, que ha sido la principal fuente de conocimiento acerca de la lyonización hasta recientemente, cuando se ha comenzado a explorar la ICX en humanos. Estas últimas investigaciones, que han sido llevadas a cabo tanto en células pluripotenciales como en embriones humanos, han permitido esclarecer algunos de los mecanismos que participan en este proceso y la relevancia que tienen en muchas enfermedades (7).
En el centro de todas las investigaciones se encuentra un gen constitutivo o estructural: el gen XIST (X Inactive Specific Transcript). Es un ARN no codificante largo (lncRNA) necesario en “cis” (8) para la inactivación del cromosoma X. La importancia de este gen radica en que se ha descrito como necesario y suficiente (4) para el proceso de Lyonización. Aunque su importancia ha ido en aumento, los últimos avances en el estudio del desarrollo embrionario y en epigenética han permitido conocer otros mecanismos distintos que participan en la ICX. Es por esta razón que el principal objetivo de este trabajo es revisar los mecanismos de inactivación del cromosoma X, centrándonos en la relevancia del gen XIST, y comentar las implicaciones de este proceso en la práctica clínica.
CUERPO DE LA REVISIÓN
1. METODOLOGÍA
Para realizar esta revisión se han utilizado artículos científicos y trabajos de investigación publicados entre 2006 y 2024, estando la mayoría de ellos comprendidos entre 2017 y 2024. También se han utilizado otros recursos bibliográficos, como vídeos de divulgación científica, capítulos de libros, páginas webs y descriptores de ciencias de la salud.
La bibliografía ha sido obtenida de Google Scholar, Pubmed, Web Of Science (WOS) y ClinicalKey.
Por la amplitud del tema a revisar y la gran cantidad de resultados que generaba la búsqueda de los tesauros en las bases de datos utilizadas, no se han establecido unos criterios de inclusión y exclusión estrictos.
2. MECANISMOS MOLECULARES
El proceso de inactivación del cromosoma X se basa en las diferencias en la carga genética entre los dos cromosomas sexuales: el X y el Y. Concretamente, el cromosoma X tiene aproximadamente 8 veces más genes que el cromosoma Y (9). Para compensar esta diferencia génica, los seres vivos presentan una serie de mecanismos moleculares que difieren entre especies.
Uno de los organismos más estudiados es el ratón.
Según las investigaciones de la doctora J. Lee et al (10), la ICX en el ratón comienza desde antes de la fecundación. En la ovogénesis, los dos cromosomas X se encuentran activos, y uno de ellos es el que se transmitirá a la descendencia. Sin embargo, durante la espermatogénesis, ambos cromosomas X e Y sufren una inactivación. Se cree que esta inactivación no solo ocurre para evitar la recombinación homóloga de ambos cromosomas, sino que podría corresponder a un mecanismo de impronta genética que silencia el cromosoma paterno en las siguientes generaciones (especialmente en las hembras) (10).
Por esta razón, en las primeras etapas del desarrollo embrionario encontramos un mecanismo de ICX en forma de impronta génica, por el que el cromosoma que siempre se encuentra inactivado es el paterno (Xp) (10). Este mecanismo se mantiene hasta la etapa de blastocisto. En esta fase del desarrollo, los tejidos extraembrionarios (p.e. la placenta) mantendrán esta forma de ICX. En cambio, en los tejidos embrionarios (el epiblasto), se llevará a cabo una reactivación de este cromosoma Xp (10). A este proceso le sigue una nueva inactivación, pero esta vez por un mecanismo distinto al de impronta: será una inactivación aleatoria del cromosoma X, en la que cualquiera de los dos puede ser inactivado (Xm o Xp) (10).
En la línea germinal del embrión femenino encontramos otro proceso de reactivación (10): el cromosoma X que fue inactivado aleatoriamente vuelve a convertirse en activo para que, en el futuro, ambos cromosomas X de la línea germinal tengan las mismas probabilidades de transmitirse a la descendencia (10).
Este mecanismo de impronta paterna en ratones, ha sido también corroborado por otras investigaciones más recientes, como las de A. Wutz (11), Khan SA (8) o S. Kalantry (12). Sin embargo, estos estudios señalan que la inactivación del Xp ocurriría tras la fecundación y que el mecanismo que marca este proceso sería el aumento de la expresión de XIST.
En humanos, el mecanismo de ICX es más complejo. El proceso de ICX podría dividirse en 3 fases: Iniciación de la ICX, establecimiento de la misma y mantenimiento (12). La iniciación se define por S. Kalantry et al (12) como la inducción de XIST a partir del cromosoma X que será inactivado. Pero para saber cuál cromosoma será inactivado, primero es necesario conocer cuántos cromosomas X hay que inactivar y cuál será el Xi. Por eso, podrían añadirse dos etapas más: la fase de conteo y la fase de selección del cromosoma Xi. Los mecanismos que subyacen a estas últimas etapas siguen siendo estudiados. Una de las hipótesis que se barajan es la teoría de los dos factores (10). En esta hipótesis, los cromosomas X tendrían un factor “numerator” (FN) y los autosomas, otro factor “denominator” (FD). Según esto, los factores se “ajustarían” mutuamente y formarían un factor bloqueador (10).
En células XY, este factor bloqueador se une al centro XIC del único cromosoma X que tienen y evita que se produzca la ICX. La región XIC o centro de inactivación del cromosoma X en inglés (13), es una zona de unas 800 kb en el brazo largo del cromosoma X (Xq13.2) que coordina la mayoría de las etapas del proceso de lyonización y contiene al principal actor de este mecanismo: el lncRNA XIST (13). Se considera necesaria y suficiente para que se produzca el proceso de ICX (14).
En células XX, se generan dos FN. Uno de ellos, se unirá como en las células XY a los FD y formará el “factor bloqueador”, el cual se unirá a uno de los dos cromosomas X, evitando así la ICX (10).
Sobre lo que ocurre con el otro cromosoma encontramos dos hipótesis (10). La primera es que el otro cromosoma es inactivado “por defecto” y la otra, que el segundo FN actuaría como “factor competencia”, situándose en XIC y, al contrario que el factor bloqueador, activando la ICX (10).
Una vez conocido el número de cromosomas X que hay que inactivar, la célula elige uno de ellos para la ICX. Sobre este proceso, el emparejamiento de ambos cromosomas X por la región Xic justo antes de la ICX fue propuesto como posible mecanismo de elección del cromosoma X a inactivar (10,15). Sin embargo, estudios más recientes (12) sostienen que este mecanismo no es tan importante para promover un aumento temprano de la expresión de XIST.
Para el estudio de los mecanismos de la ICX, podemos dividir el proceso en dos períodos: el período preimplantacional y el postimplantacional (8).
2.1. ICX EN PERIODO PREIMPANTACIONAL
Las diferencias en los mecanismos de compensación de dosis génica entre humanos y modelos murinos son más notables en el período preimplantacional (Tabla 1)(8).
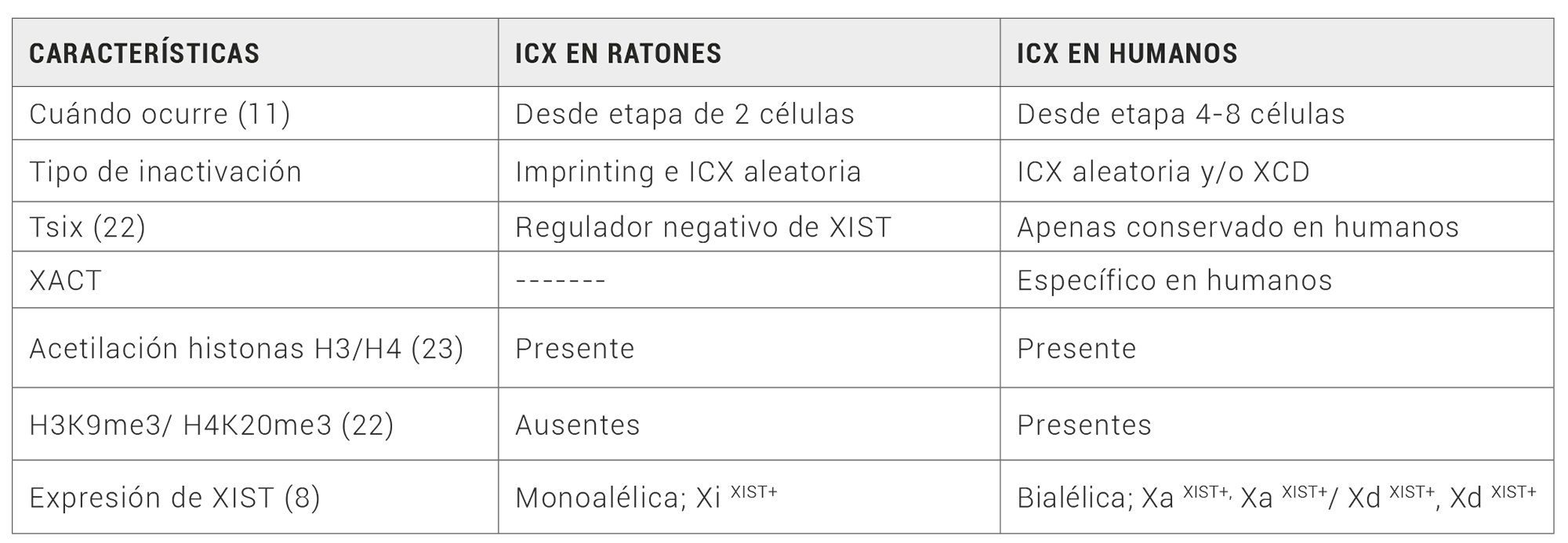
Tabla 1. Tabla resumen de las diferencias en el proceso de inactivación del cromosoma X entre ratones y humanos (8, 11, 22, 23)
Esta etapa se limita hasta el E4.5-4.75 (“Embryonic days”) en modelos murinos y hasta E7-8 en humanos, que corresponden a los días en los que se produce la implantación (16). Es en este periodo en el que se producen la mayoría de los procesos relacionados con la ICX.
La complejidad del proceso de lyonización en humanos imposibilita dilucidar los mecanismos exactos por los que éste se rige, estableciéndose numerosas teorías al respecto. Una de estas hipótesis es la llamada “X chromosome dampening (XCD)”, propuesta por Petropoulos et al (17). Según esta teoría (17), la ICX no ocurriría de manera similar a como ocurre en los ratones, en los que, cómo se explicó anteriormente, uno de los dos cromosomas X es inactivado. En el proceso de XCD, se describe cómo ambos cromosomas X sufren una disminución parcial de su expresión genética al inicio del desarrollo embrionario, siendo XIST el principal candidato para mediar este mecanismo.
Otros autores, como Khan (8) o Plath (18,19) apoyan el proceso de XCD. Sin embargo, otros investigadores como Moreira de Mello et al (20) y Collombet et al (14) discuten la existencia de este mecanismo de XCD, explicando que durante el desarrollo embrionario ocurre una disminución de la expresión de genes bialélicos asociados al cromosoma X, lo cual es más compatible con el proceso de inactivación del cromosoma X aleatoria (ICXa/ en inglés: Rxci).
Según este modelo de ICX aleatorio, XIST no es el único actor en el proceso de Lyonización: hay varias proteínas y lncRNAs que participan en este fenómeno, además de otros cambios epigenéticos como modificaciones de histonas.
En modelos murinos, el lncRNA Tsix ha sido reconocido como represor de XIST, sin embargo, se cree que no existe un equivalente de este en humanos (7), ya que su locus se encuentra pobremente conservado (8). Uno de los RNA que sí que ha sido sugerido como regulador de XIST en humanos es el X-active coating transcript (XACT). Se co-acumula con XIST en los cromosomas X y parece que controla la asociación de XIST al cromosoma en cis (21), además de alterar la habilidad de éste de inactivar el cromosoma X (14). Otros reguladores que son comunes en ratones y humanos son dos genes de la región XIC: Ftx y Jpx. Ambos contribuyen a la expresión de XIST y uno de ellos, el gen Jpx, es considerado el primer gen que se activa dentro de la región XIC, incluso antes que el propio XIST (14).
A nivel epigenético, destacan modificaciones en histonas, como por ejemplo la trimetilación de H3K27 (H3K27me3) o la inhibición de la subunidad EZH2 del Polycomb Repressive Complex 2 (PRC2). Aunque han sido ampliamente estudiadas, se ha visto que son dispensables para la expresión de XIST y el proceso de ICX (8,11).
2.2. ICX EN PERIODO POSTIMPLANTACIONAL
Aunque las investigaciones acerca del proceso de ICX se centren en estudiar los mecanismos moleculares antes de la implantación, algunos autores consideran que la ICX como tal no ocurre hasta un poco después de ésta (7,17).
Una de las proteínas más recientemente estudiadas que parece participar en el establecimiento del cromosoma X inactivo es SPEN (12), aunque la mayoría de los estudios han sido realizados en modelos murinos. Por último, destacar el papel de las proteínas PRC1 y PRC2, así como de SMCHD1, cuya función en el mantenimiento del cromosoma Xi ha sido estudiada sobre todo en modelos murinos (12).
3. IMPORTANCIA DE LA ICX EN LA PRÁCTICA CLÍNICA
Entre todas las líneas de estudio sobre este tema, son tres las que se van a desarrollar en este apartado: las enfermedades ligadas al cromosoma X, la relación de la ICX con las enfermedades autoinmunes y con el cáncer.
3.1. ENFERMEDADES LIGADAS AL CROMOSOMA X
Algunas enfermedades, como la distrofia muscular de Duchenne o la incontinencia pigmenti son enfermedades que presentan un patrón de herencia ligado al cromosoma X. De forma general, podemos decir que en casi todos los trastornos que se transmiten ligados al cromosoma X, la enfermedad es más grave en varones que en mujeres. Esto se debe al mecanismo de ICX: los hombres suelen ser hemicigotos para los genes relacionados con el cromosoma X (3), por lo que, al mantener activo el único cromosoma X que tienen, expresan la enfermedad con toda la clínica asociada. Sin embargo, las mujeres presentan dos tipos celulares según el proceso de ICX: las que tienen el cromosoma X materno inactivo y las que tienen el paterno inactivo. Por lo tanto, solo una proporción de células presentará el alelo mutado inactivado, haciendo que las mujeres presenten una clínica más leve de la enfermedad (23).
Este mecanismo de transmisión del material genético puede dividirse a su vez en dos tipos: herencia recesiva y herencia dominante ligada al X. (Figura 1)
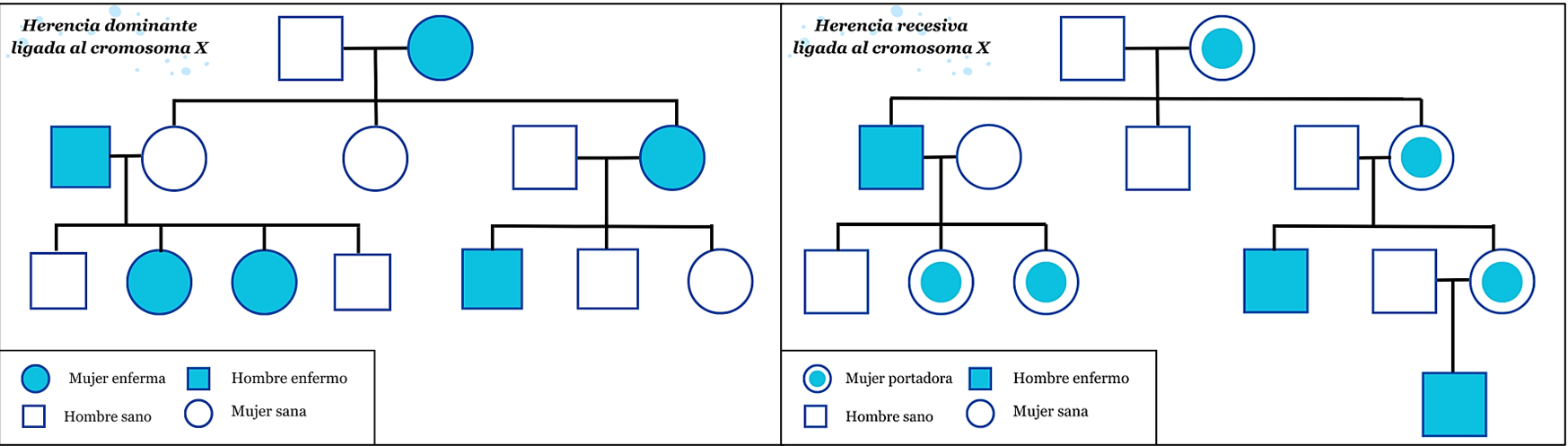
Figura 1. Árboles genealógicos de los dos tipos de herencia ligada al cromosoma X. A la derecha: herencia recesiva ligada al cromosoma X. A la izquierda: herencia dominante ligada al cromosoma X. Se observa como en la herencia dominante ligada al cromosoma X las mujeres se ven afectadas aproximadamente el doble que los hombres (2 varones enfermos y 4 mujeres enfermas en el árbol). Sin embargo, en la herencia recesiva vemos como las mujeres son portadoras, siendo los afectados los varones (3,24).
En la tabla 2 se pueden ver las diferencias entre sexos según el tipo de herencia: cómo mientras que la afectación es más frecuente en hombres en la enfermedad recesiva ligada al cromosoma X, en las enfermedades dominantes ligadas al X, las mujeres son las que se ven más afectadas (3,25).
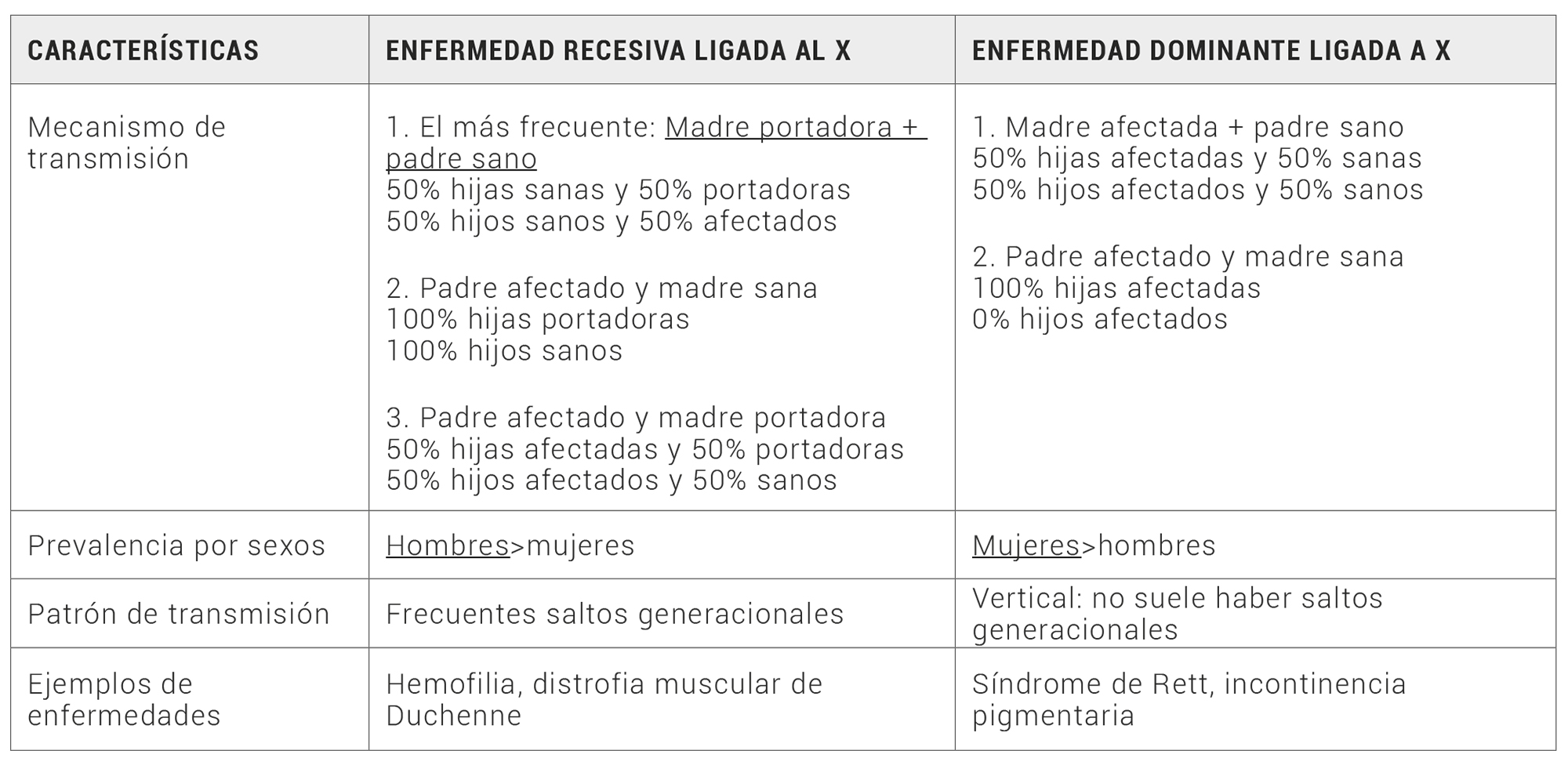
Tabla 2. Características de los tipos de herencia ligada al cromosoma X (3, 25)
Esto se relaciona con la ya mencionada diferencia en la carga genética entre hombres y mujeres; mientras que los hombres solo presentan una copia del cromosoma X (25), las mujeres tienen dos (3). Por este motivo, en las enfermedades recesivas ligadas al X, si el hombre hereda un alelo mutado del cromosoma X, expresará la enfermedad, pues el cromosoma Y no tiene una copia normal para compensar al patológico. Por tanto, un hombre solo podrá ser enfermo o sano con respecto a una enfermedad recesiva ligada a X. Sin embargo, en las mujeres, pueden ocurrir tres situaciones (25): que la mujer reciba dos cromosomas X con la mutación (homocigota para el alelo mutado), que reciba dos cromosomas X sin la mutación (homocigota para el alelo natural) o que reciba solo un cromosoma X mutado (heterocigota para la mutación). En el primer caso, la mujer sería enferma, en el segundo caso, sana, y en el último caso, se consideraría portadora de la mutación. Las portadoras pueden tener algunos síntomas, aunque no expresan todo el fenotipo de la enfermedad, ya que, por el proceso de inactivación del cromosoma X, solo la mitad de sus células expresarán el alelo mutado (25).
Otro caso distinto es el de la herencia dominante ligada al cromosoma X. En este caso, solamente tiene que heredarse un alelo mutado para expresar la enfermedad. Al tener las mujeres dos copias del cromosoma X, pudiendo tener cada una el alelo mutado, la frecuencia de mujeres afectadas es el doble que en varones, por regla general. Excepciones a esta norma serían enfermedades que sean mortales en los hombres, como por ejemplo el síndrome de Rett y la Incontinencia pigmentaria (2).
La incontinencia pigmentaria/ Síndrome de Bloch-Siemens es una displasia ectodérmica (26) con herencia dominante ligada al cromosoma X que afecta casi exclusivamente a mujeres, con una gran variabilidad clínica y siendo letal para la mayoría de los varones intraútero (27). La clínica cutánea aparece desde el nacimiento y se constituye de cuatro fases no secuenciales: una fase ampollosa, una fase verrugosa, una fase de hiperpigmentación y una fase atrófica (26). También presentan anomalías neurooftalmológicas, alteraciones dentarias, alopecia y distrofia ungueal (26).
La gran variabilidad clínica descrita en mujeres es atribuida al fenómeno de ICX (27). En los casos de Incontinencia pigmentaria en varones, hay que estudiar tres mecanismos por los cuales ha sido posible la supervivencia de estos: mosaicismo somático, mutaciones menos agresivas que provoquen otros fenotipos, o que presente un exceso de cromosomas X (como por ejemplo un Síndrome de Klinefelter, 47XXY) (27). Este último mecanismo vuelve a llevarnos al proceso de Lyonización: al inactivarse uno de los dos cromosomas X, algunas células quedarían con el cromosoma X mutado, mientras que otras tendrían el alelo natural, como ocurre en las mujeres.
3.2. AUTOINMUNIDAD E INACTIVACIÓN DEL CROMOSOMA X
Las enfermedades autoinmunes sistémicas (EAS) son un grupo de enfermedades que pueden afectar a varios órganos y sistemas y cuyo origen es autoinmunitario (28,29). Estas enfermedades predominan en el sexo femenino, pues se ha visto que de un 4% de la población (30) que padece una enfermedad autoinmune, el 75% son mujeres (30).
Aunque las hormonas han sido ampliamente estudiadas como el principal desencadenante de esta predisposición en las mujeres a las EAS, estudios recientes señalan al cromosoma X como uno de los principales promotores de autoinmunidad (31,32). En concreto, esta asociación parece estar relacionada con el escape de ciertos genes del proceso de ICX (23) y con el exceso de cromosomas X (23,31).
La ICX es un proceso incompleto: entre un 15 y un 23% de los genes del cromosoma X escapan al proceso de lyonización (33). Un porcentaje de estos genes corresponde a las regiones pseudoautosómicas del cromosoma X (PAR), que presentan regiones homólogas en el cromosoma Y. (33) Sin embargo, el resto de genes que escapan al proceso de lyonización son propios del cromosoma X, el cual contiene una gran cantidad de genes relacionados con la respuesta inmunitaria (33). Estas regiones que escapan al mecanismo de ICX se expresarían tanto por el cromosoma X inactivo (Xi) como por el activo (Xa) (33), por lo que, al ser transcritos en base a dos alelos en las hembras, podrían generar diferencias tanto en la cantidad como en la actividad de las proteínas en las mujeres con respecto a los hombres (34).
Los genes que escapan al proceso de ICX pueden considerarse de dos tipos: genes que escapan a la ICX constitutivos y genes que escapan a la ICX facultativos (35). Son éstos últimos los de mayor relevancia a nivel de autoinmunidad, pues su escape del proceso de lyonización es variable entre células, tejidos e incluso personas (35), además son los que más se han asociado con procesos autoinmunes, particularmente al Lupus Eritematoso Sistémico (LES). Algunos de los genes que más se han relacionado son CD40L, CXCR3, BTK, IRAK-1, TLR7/TLR8 o CXorf21 (33,35).
Esta teoría se reafirma con las observaciones en síndromes que presentan un exceso de cromosomas X: los pacientes con síndrome de Klinefelter presentan igual (36) o incluso hasta 15 a 25 veces más riesgo de sufrir enfermedades como LES (14) o Síndrome de Sjögren y las mujeres con el síndrome de triple X (47,XXX), presentan igualmente un riesgo aumentado de sufrir LES (14).
Recientemente se ha relacionado la prevalencia de procesos autoinmunes en mujeres con uno de los pilares principales del proceso de ICX: el lncRNA XIST. El estudio de Howard Y Chang et al (31) investigaba por primera vez la inmunogenicidad del propio complejo XIST RNP (31). La investigación fue llevada a cabo en ratones y la enfermedad objeto de estudio fue el LES. Se observó que la expresión de este complejo en ratones macho aumentaba la severidad de la enfermedad y promovía cambios en el perfil de las células B y T que están implicadas en la patogénesis del LES (31). Por tanto, los autores (31) proponen al complejo XIST como uno de los desencadenantes antigénicos que podría explicar la mayor prevalencia de enfermedades autoinmunes en mujeres que en hombres.
El papel de XIST también ha sido señalado como relevante en otro ámbito: en la patogénesis del cáncer.
3.3. ASOCIACIÓN DEL PROCESO DE LYONIZACIÓN Y CÁNCER
Aunque el papel de XIST siempre ha sido estudiado en el período de desarrollo embrionario, investigaciones recientes (37) sostienen la teoría de que fluctuaciones de los niveles de XIST podrían regular la expresión de los genes que escapan del proceso de ICX y, en algunos tipos celulares, asociarse a cáncer (37). Algunos estudios, tanto en humanos como en modelos murinos, han observado que la pérdida/eliminación de XIST en algunos tejidos (en concreto, en tejido mamario y hematopoyético) promueve la aparición de cáncer. En el estudio de C.Ginestier et al (38), observaron una pérdida de XIST en todos los tipos moleculares de tumores mamarios, siendo especialmente prevalente esta pérdida en los más agresivos (38). En la investigación de JT Lee et al (10, 39), la deleción de XIST indujo una neoplasia hematológica con penetrancia completa en hembras.
La relación del cáncer con la ICX no solo depende de XIST; algunos de los genes que escapan del proceso de ICX están relacionados con el proceso de tumorigenésis. Estos son conocidos como “escape from X-inactivation tumor suppressor (EXITS) genes” (40). Al tener dos cromosomas X y al escapar los genes EXITS del proceso de ICX, las mujeres presentan dos copias activas de éstos, al contrario que los hombres, que al tener un único cromosoma X sólo presentan una copia (40). Esta expresión bialélica de estos genes en mujeres las protege de las mutaciones con pérdida de función, explicándose así la mayor incidencia de distintos tipos de cáncer en los hombres (14). Son seis genes en los que se ha observado mayor frecuencia de mutaciones: ATRX, CNKSR2, DDX3X, KDM5C, KDM6A, and MAGEC3 (40).
DISCUSIÓN
Nuestro conocimiento acerca del proceso de inactivación del cromosoma X ha crecido exponencialmente (8) desde que Mary Lyon propuso el concepto de lyonización en 1961.
Las investigaciones llevadas a cabo con embriones humanos procedentes de fecundación en vitro (FIV) han permitido comprender mejor cómo funciona el proceso de ICX en humanos (7), aunque parece haber algunas limitaciones. Catherine Patrat et al (7) señalan cómo muchos de los embriones de técnicas FIV no presentan un proceso de desarrollo típico, ya que una gran parte de ellos son concebidos en contextos de infertilidad. Además, si el proceso de ICX ya se ha establecido en el embrión en estudio, éste mantendrá la inactivación, imposibilitando analizar los mecanismos subyacentes (8).
Por estas razones, en estudios más recientes han comenzado a utilizarse células pluripotentes humanas (hPSCs, en inglés), que permiten el estudio ex vivo (7) de las primeras etapas del desarrollo embrionario. Éstas pueden ser de dos tipos: células madre pluripotenciales embrionarias y células madre pluripotenciales inducidas (7,41). Un tipo específico de estas células, las hPSCs naïve, son capaces de expresar la ICX de novo, a diferencia de lo que ocurría en los estudios con embriones (8).
Estas investigaciones en humanos han permitido observar las diferencias existentes con la ICX en ratones, además de plantear hipótesis acerca de algunos mecanismos relacionados con la lyonización que no han sido observados en ratones, como por ejemplo son el proceso de “Dampening” (XCD) o XACT.
CONCLUSIONES
El reciente inicio de las investigaciones en humanos sobre la ICX ha abierto las puertas hacia el conocimiento de unos mecanismos mucho más desconocidos de lo que se pensaba. Aun siendo recientes, ya se ha visto que hay más diferencias de las esperadas en el proceso de ICX entre ratones y humanos (7). Conociendo cómo funcionan los mecanismos que permiten la inactivación del cromosoma X, se podrá entender qué relevancia tiene este proceso en muchas patologías, como por ejemplo el LES o el cáncer. Igualmente, conocer qué moléculas actúan en la ICX podrá permitirnos actuar frente a ciertas enfermedades. Algunos estudios (40,42) ya han comenzado a investigar las posibles aplicaciones en la práctica clínica del principal actor en la ICX: el lncRNA XIST. Éste ha sido estudiado tanto como tratamiento para algunos tumores (40) como en el síndrome de Rett, aumentando la expresión de MECP2 usando un oligonucleótido antisentido de XIST (42).
Esto es solo el comienzo de una época en la que la genética va a participar activamente en el manejo de las enfermedades en la práctica clínica, siendo el proceso de ICX una pequeña parcela dentro de ésta.
AGRADECIMIENTOS
Al Dr. M. Alaminos, por haberme guiado y aconsejado sobre la elaboración de este artículo.
REFERENCIAS bibliográficas
- ↑Inactivación del cromosoma X [Internet]. DeCS – Descriptores en ciencias de la salud. 2024 [citado 6 de febrero de 2024]. Recuperado a partir de: https://decs.bvsalud.org/es/ths/resource/?id=51238&filter=ths_termall&q=Inactivaci%C3%B3n%20cromosoma%20X#Concepts
- ↑Migeon BR, Beer MA, Bjornsson HT. Embryonic loss of human females with partial trisomy 19 identifies region critical for the single active X. PLoS One 2017 Apr 12;12(4):e0170403.
- ↑Jorde, Lynn B., PhD; Carey, John C., MD, MPH; Bamshad, Michael J., MD. Modos de herencia ligados al sexo y no clásicos – Genética médica. En: Jorde, Lynn B., PhD. Genética médica 6ª edición. Elsevier España; 2021. p.73-96.
- ↑Ahn, J. & Lee, J. (2008) X chromosome: X inactivation. Nature Education 1(1):24
- ↑Liu J, Ali M, Zhou Q. Establishment and evolution of heterochromatin. Ann N Y Acad Sci 2020;1476(1):59-77.
- ↑Balaton BP, Brown CJ. Contribution of genetic and epigenetic changes to escape from X-chromosome inactivation. Epigenetics & Chromatin 2021;14(1):30.
- ↑Patrat C, Ouimette J, Rougeulle C. X chromosome inactivation in human development. Development 2020;147(1):dev183095)
- ↑Khan SA, Theunissen TW. Modeling X-chromosome inactivation and reactivation during human development. Curr Opin Genet Dev 2023 Oct;82:102096.
- ↑McIntyre KL, Waters SA, Waters PD. Sex Chromosomes. En: Skinner MK, editor. Encyclopedia of Reproduction (Second Edition) Oxford: Academic Press; 2018. p. 238-244.
- ↑Science Communication Lab. Jeannie Lee (Harvard) 1- X Chromosome Inactivation: Making and Breaking the Silence [vídeo]. 8 de mayo de 2019. [1 de abril de 2024]. Disponible en: https://www.youtube.com/watch?v=y3ST0whbA4k
- ↑Kaufmann C, Wutz A. IndiSPENsable for X Chromosome Inactivation and Gene Silencing. Epigenomes 2023;7(4).
- ↑Malcore RM, Kalantry S. A Comparative Analysis of Mouse Imprinted and Random X-Chromosome Inactivation. Epigenomes 2024 Feb 10;8(1):8. doi: 10.3390/epigenomes8010008.
- ↑Nussbaum RL, McInnes RR, Willard HF. Bases cromosómicas y genómicas de la enfermedad: trastornos de los autosomas y de los cromosomas sexuales. En: Nussbaum RL., McInnes RR., Willard HF. Thompson & Thompson. Genética en Medicina [Internet]; 7ª edición. Elsevier España; 2016. [citado abril 2024] p. 75-105.
- ↑Loda A, Collombet S, Heard E. Gene regulation in time and space during X-chromosome inactivation. Nature Reviews Molecular Cell Biology 2022;23(4):231-249
- ↑Xu N, Tsai C, Lee JT. Transient homologous chromosome pairing marks the onset of X inactivation. Science 2006 Feb 24;311(5764):1149-1152.
- ↑Molè MA, Weberling A, Zernicka-Goetz M. Chapter Four – Comparative analysis of human and mouse development: From zygote to pre-gastrulation. Curr Top Dev Biol 2020;136:113-138.
- ↑Petropoulos S, Edsgärd D, Reinius B, Deng Q, Panula SP, Codeluppi S, et al. Single-Cell RNA-Seq Reveals Lineage and X Chromosome Dynamics in Human Preimplantation Embryos. Cell 2016;165(4):1012-1026.
- ↑Sahakyan A, Kim R, Chronis C, Sabri S, Bonora G, Theunissen TW, et al. Human naive pluripotent stem cells model X chromosome dampening and X inactivation. Cell stem cell 2017;20(1):87-101
- ↑Dror I, Chitiashvili T, Tan SYX, Cano CT, Sahakyan A, Markaki Y, et al. XIST directly regulates X-linked and autosomal genes in naive human pluripotent cells. Cell 2024;187(1):110-129.e31
- ↑Moreira de Mello JC, Fernandes GR, Vibranovski MD, Pereira LV. Early X chromosome inactivation during human preimplantation development revealed by single-cell RNA-sequencing. Scientific Reports 2017;7(1):10794
- ↑Vallot C, Patrat C, Collier AJ, Huret C, Casanova M, Liyakat Ali TM, et al. XACT Noncoding RNA Competes with XIST in the Control of X Chromosome Activity during Human Early Development. Cell Stem Cell 2017;20(1):102-111
- Sado T, Sakaguchi T. Species-specific differences in X chromosome inactivation in mammals. Reproduction 2013;146(4):R131-R139.
- ↑Fan J, Jin X, Sun Z, Wang Y. X-Chromosome Inactivation and Related Diseases. Genetics Research 2022;2022:e67.
- Frost Amy, Tatton-Brown. X-linked inheritance [Internet]. NHS England. National Genomics Education Program / GeNotes. 2022 [citado el 21 de abril de 2024]. Recuperado a partir de: https://www.genomicseducation.hee.nhs.uk/genotes/knowledge-hub/x-linked-recessive-inheritance/
- ↑Giraldo Garcia, AM, Castaño Osorio J.C. Herencia ligada al cromosoma X. En: Gómez Marín J.E. González Marín Á. y Castaño Osorio J.C. Fundamentos básicos de medicina: biología molecular: principios y aplicaciones [En Línea]. Medellín: Fondo Editorial CIB, 2011 [consultado 20 Abril 2024]. P 243-256. Disponible en: https://elibro.net/es/lc/ugr/titulos/186737
- ↑Scheurle Angela. Incontinencia pigmentaria [Internet]. Enciclopedia de Orphanet. 2019 [citado 21 de abril de 2024]. Recuperado a partir de: https://www.orpha.net/es/disease/detail/464?name=Incontinencia%20pigmentaria&mode=name
- ↑Feito-Rodríguez M, García-Macarrón J, Ruiz Bravo-Burguillos E, Vera-Casaño Á, de Lucas-Laguna R. Incontinentia pigmenti: tres nuevos casos que demuestran que no es sólo cosa de mujeres. Actas Dermo-Sifiliográficas 2007;98(2):112-115.
- ↑Enfermedades reumáticas autoinmunes sistémicas. [Internet]. SARE Sociedad Asturiana de Reumatología. 2017. Recuperado a partir de: https://sare.org.es/enfermedades-reumaticas-autoinmunes-sistemicas/
- ↑Enfermedades autoinmunes sistémicas (EAS). [Internet] Fundación Española de Reumatología. Recuperado a partir de: https://inforeuma.com/enfermedades-reumaticas/enfermedades-autoinmunes-sistemicas-eas/
- ↑El 4% de la población padece una enfermedad autoinmune sistémica y el 75% de afectados son mujeres. [Internet]. Sociedad Española de Reumatología. 19 de febrero de 2024. Recuperado a partir de: https://www.ser.es/el-4-de-la-poblacion-padece-una-enfermedad-autoinmune-sistemica-y-el-75-de-afectados-son-mujeres/
- ↑Dou DR, Zhao Y, Belk JA, Zhao Y, Casey KM, Chen DC, et al. Xist ribonucleoproteins promote female sex-biased autoimmunity. Cell 2024;187(3):733-749.e16.
- ↑Anesi N, Miquel C, Laffont S, Guéry J. The Influence of Sex Hormones and X Chromosome in Immune Responses. In: Klein SL, Roberts CW, editors. Sex and Gender Differences in Infection and Treatments for Infectious Diseases Cham: Springer International Publishing; 2023. p. 21-59
- ↑Youness A, Miquel C, Guéry J. Escape from X Chromosome Inactivation and the Female Predominance in Autoimmune Diseases. International Journal of Molecular Sciences 2021;22(3).
- ↑Gal-Oz S, Shay T. Genetics of Sex Differences in Immunity. In: Klein SL, Roberts CW, editors. Sex and Gender Differences in Infection and Treatments for Infectious Diseases Cham: Springer International Publishing; 2023. p. 1-19.
- ↑Miquel C, Faz-Lopez B, Guéry J. Influence of X chromosome in sex-biased autoimmune diseases. J Autoimmun 2023;137:102992.
- ↑Sierra I, Anguera MC. Enjoy the silence: X-chromosome inactivation diversity in somatic cells. Curr Opin Genet Dev 2019;55:26-31.
- ↑Antonia Hauth, Jasper Panten, Emma Kneuss, Christel Picard, Nicolas Servant, Isabell Rall, et al. Escape from X inactivation is directly modulated by levels of Xist non-coding RNA. bioRxiv 2024:2024.02.22.581559 [Preprint]. 2024 [citado 24 de abril de 2024]. doi: 10.1101/2024.02.22.581559
- ↑Richart L, Picod-Chedotel M, Wassef M, Macario M, Aflaki S, Salvador MA, et al. XIST loss impairs mammary stem cell differentiation and increases tumorigenicity through Mediator hyperactivation. Cell 2022;185(12):2164-2183.e25.
- ↑Yildirim E, Kirby JE, Brown DE, Mercier FE, Sadreyev RI, Scadden DT, et al. Xist RNA Is a Potent Suppressor of Hematologic Cancer in Mice. Cell 2013;152(4):727-742.
- ↑Wang D, Tang L, Wu Y, Fan C, Zhang S, Xiang B, et al. Abnormal X chromosome inactivation and tumor development. Cellular and Molecular Life Sciences 2020;77(15):2949-2958.
- ↑Pluripotent stem cell [Internet]. Nature portfolio- Nature. 2024. [Citado 2 de mayo de 2024]. Recuperado a partir de: https://www.nature.com/subjects/pluripotent-stem-cells
- ↑Carrette LLG, Wang C, Wei C, Press W, Ma W, Kelleher RJ, et al. A mixed modality approach towards Xi reactivation for Rett syndrome and other X-linked disorders. Proceedings of the National Academy of Sciences 2018;115(4):E668-E675.
INFORMACIÓN DEL ARTÍCULO
Conflicto de intereses: Los autores de este artículo declaran no tener ningún tipo de conflicto de intereses respecto a lo expuesto en el presente trabajo.
Correspondencia: Alba Martín Hernández. Facultad de Medicina de la Universidad de Granada, Avda. de la Investigación 11, 18016. E-mail: albamartinh@correo.ugr.es





