

Caso Clínico
Disección aórtica en paciente con mutación del gen ACTA2: a propósito de un caso
Aortic dissection caused by the mutation of the ACTA2 gene: report of a case
Actual Med.2025;110(821):46-50 DOI: 10.15568/am.2025.821.cc02
Recibido: 02/02/2022
Revisado: 19/02/2022
Aceptado: 05/12/2024
RESUMEN
Las mutaciones en el gen ACTA2 producen alteraciones en la contractilidad del musculo liso que pueden manifestarse, en muchas ocasiones, como enfermedad aórtica torácica con herencia autosómica dominante. La disección aórtica es una entidad clínica con alta mortalidad que requiere de un diagnóstico y un tratamiento precoz. En algunos casos, el análisis genético juega un papel decisivo. Exponemos el caso clínico de una mujer de 45 años con mutación en una variante del gen ACTA2, que presenta una disección aórtica espontánea asociada a un accidente isquémico transitorio.
Palabras clave: ACTA2; Disección aórtica; Arteriopatía; Alfa-actina; Músculo liso; Ictus.
ABSTRACT
Alterations in smooth muscle contractility can be produced by mutations in the ACTA2 gene. This autosomal dominant inheritance disease can be manifested as a thoracic aortic dissection. Most of the time, early diagnosis and treatment are required due to the high mortality of this clinical entity. In some cases, genetic analysis plays a decisive role. We expose the clinical case of a 45-year-old woman with a mutation in a variant of the ACTA2 gene, who presents a spontaneous aortic dissection associated with a transient ischemic attack.
Keywords: ACTA2; Aortic dissection; Arteriopathy; Alpha-actin; Smooth muscle; Stroke
Leer Artículo Completo
Introducción
La disección aórtica aguda tipo A (DAATA) se produce por un desgarro de las capas que conforman la pared arterial de la aorta ascendente. Esta entidad clínica presenta altas tasas de mortalidad operatoria, entre un 20-25% de promedio. Sin embargo, este porcentaje se ve considerablemente aumentado en pacientes que presenten mala perfusión mesentérica asociado a ictus y/o unos niveles de lactato sérico ≥ 6 mmol / L (1). Además, el 10% de los pacientes con DAATA requieren una reintervención quirúrgica que puede presentar alta tasa de complicaciones por la dificultad de la técnica (2). Debido a todo lo expuesto anteriormente, es fundamental realizar un diagnóstico y abordaje precoz. Entre los factores de riesgo que propician esta patología se encuentran la edad avanzada, la hipertensión, la aterosclerosis, el tabaquismo, enfermedades inflamatorias de las arterias como arteritis o vasculitis, y enfermedades hereditarias del tejido conectivo como el síndrome de Marfan o el de Ehlers-Danlos, entre ellos. Este último grupo es especialmente vulnerable por las alteraciones genéticas que predisponen a una mayor debilidad parietal de las arterias.
Las fibras de actina junto a miosina, desmina y vimentina, conforman la maquinaria contráctil de las células del músculo liso presente en la pared arterial. Estas participan en la generación de la fuerza muscular, la estructura, la integridad y la señalización intercelular. Por este motivo, las mutaciones genéticas que afecten a estas fibras pueden debilitar la pared arterial de grandes vasos. Se han descrito algunas alteraciones genéticas que se deben tener en cuenta como MYH11, ACTA2, MMP2, MMP9, MYLK o CREBBP (3,4).
Presentamos el caso de una paciente de 45 años que acude al Servicio de Urgencias por dolor epigástrico irradiado a la espalda, parálisis facial y pérdida de visión autolimitada. Se le realiza una angioTC que revela la presencia de una DAATA con afectación de las ramas del arco aórtico. Es intervenida y en el seguimiento clínico tras el alta se le diagnostica de una mutación en el gen ACTA2.
Caso clínico
Mujer de 45 años que acude al Servicio de Urgencias sin antecedentes familiares de interés y personales de valvulopatía, intervenida a los 3 años de edad, y presencia de hernias discales cervicales, dispepsia y colelitiasis asintomática. Refiere dolor y ardor epigástrico irradiado a espalda con parálisis facial central y pérdida de visión autolimitada. Tras 48 horas, vuelve al Servicio de Urgencias con una presentación clínica similar y se le realiza un ECG que revela una elevación del segmento ST menor de 1 mm en cara inferior con elevación de la troponina I y proteína C reactiva. Ante la sospecha de patología aórtica se le realizo una angiografía por tomografía computarizada (angioTC) que reveló una dilatación aneurismática de 6 cm de diámetro asociada a hematoma intramural del cayado aórtico y disección tipo A de Stanford con afectación de la raíz y cayado aórticos y extensión al tronco braquiocefálico, arteria subclavia derecha y ostium de la arteria subclavia izquierda, junto con derrame pericárdico moderado (Figura 1). Además, como hallazgo incidental se objetivaron dos vértebras torácicas “en mariposa”.
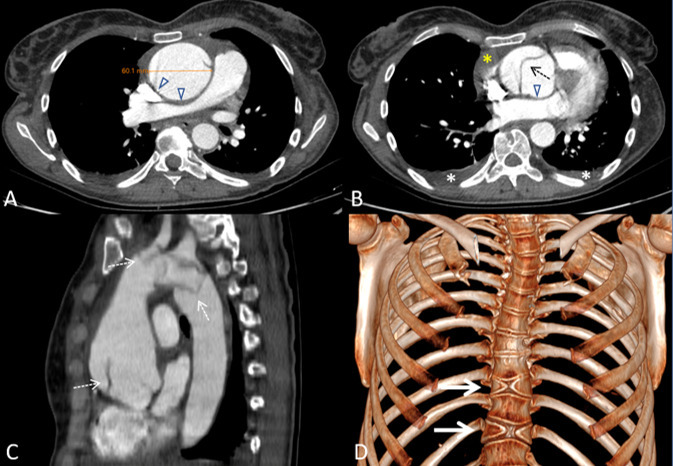
Figura 1. Angiografía por tomografía computarizada para estudio de arteria aorta y sus ramas. (A) Imagen axial a nivel del tronco de la arteria pulmonar. Dilatación de la aorta ascendente que alcanza un calibre máximo de 6,1 cm y comprime la arteria pulmonar principal derecha y la vena cava superior (puntas de flecha). (B) Imagen axial a nivel de la raíz aórtica. Extenso defecto de repleción lineal en la luz de la raíz aórtica compatible con flap intimal (flecha discontinua). Apréciese el derrame pericárdico (asterisco amarillo) y el derrame pleural bilateral en cuantía leve (asteriscos blancos). (C) Reconstrucción multiplanar, vista sagital oblicua. Dilatación de la raíz aórtica y aorta ascendente con flap intimal irregular con extensión a troncos supraaórticos (flechas discontinuas). (D) Reconstrucción volumétrica para visualización ósea. Se observan dos defectos de fusión vertebral (“vértebras en mariposa”) a nivel de T9 y T11 (flechas).
Se intervino quirúrgicamente con sustitución de aorta ascendente, cayado aórtico y una porción de la aorta descendente por prótesis Thoraflex y reimplante de los troncos supraaórticos. Tras la intervención, se realizó una ecocardiografía transtorácica que no evidenció alteraciones en las cavidades cardiacas ni valvulares ni derrame pericárdico, únicamente la raíz aórtica mostró signos de dilatación moderada. El estudio postoperatorio se completó con una angioTC que objetivó la permeabilidad de los troncos supraaórticos (Figura 2).
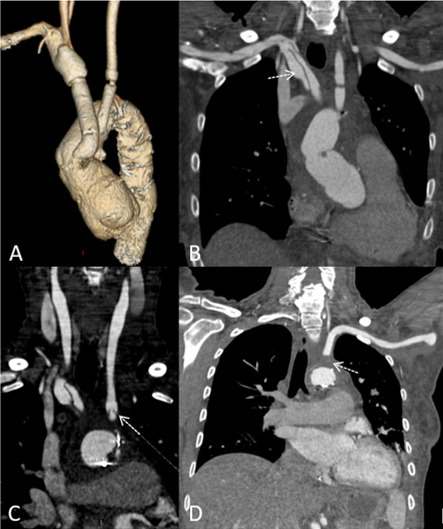
Figura 2. AngioTC de troncos supraaórticos. Reconstrucción volumétrica (A) y reconstrucciones multiplanares en plano coronal a nivel del tronco braquiocefálico derecho (B), carótida común izquierda (C) y subclavia izquierda (D). Cambios postquirúrgicos tras intervención por disección de aorta tipo A con sustitución de aorta ascendente y parte de descendente por prótesis Thoraflex y reimplante de troncos supraaórticos (A). Disección que afecta a trayecto proximal del tronco braquiocefálico derecho distal a la unión con la prótesis Thoraflex (flecha en B). Trombosis del origen de la arteria subclavia izquierda (flecha en C), con repermeabilización 2 cm distal a la oclusión. Estenosis de aproximadamente el 75% a nivel de la anastomosis de la pr ótesis Thoraflex a la arteria carótida común izquierda (flecha en D).
La paciente evolucionó favorablemente durante la recuperación postquirúrgica, manteniendo una función hemodinámica y renal adecuada, y no volvió a presentar sintomatología neurológica. Fue dada de alta con doble antiagregación durante cuatro semanas. Debido a la sospecha diagnóstica de una posible causa genética se solicita un análisis clínico que da positivo para una variante del gen ACTA2, c. 773G>A, p. (Arg258His), en el exón 7, de sentido erróneo y de herencia autosómica dominante. A los 6 meses de seguimiento, se le realizó una prueba de imagen por resonancia magnética que puso de manifiesto lesiones residuales microembólicas tanto supra como infrantentoriales de distribución corticosubcortical (Figura 3).
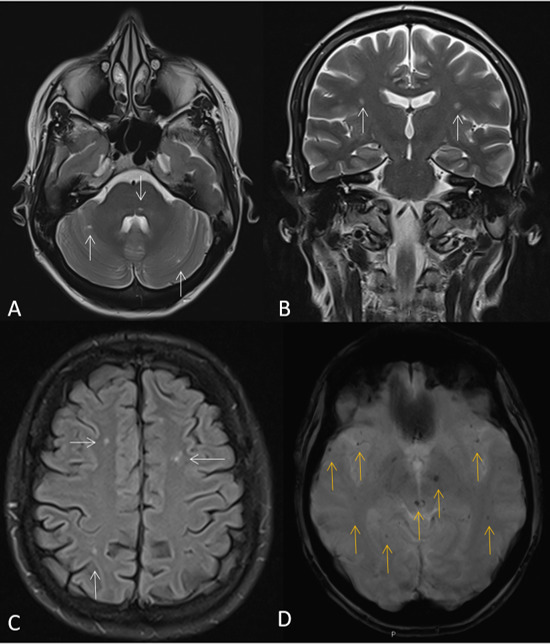
Figura 3. Resonancia magnética craneal sin contraste intravenoso realizada durante el seguimiento de la paciente, 6 meses tras el episodio agudo. Imágenes axiales potenciadas en T2 TSE (A), FLAIR (C) y T2* (reconstrucción minIP, D), e imagen coronal potenciada en T2 (B). Se observan lesiones puntiformes hiperintensas en secuencias T2 y FLAIR a nivel infratentorial (protuberancia y ambos hemisferios cerebelosos, flechas en A) y supratentorial (ambas coronas radiatas, flechas en B; y ambos centros semiovales, flechas en C) en relación con lesiones isquémicas cuyo patrón de distribución es sugerente de origen embólico. En la secuencia T2* se observan múltiples lesiones puntiformes hipointensas compatibles con focos microhemorrágicos (depósitos de hemosiderina), de distribución corticosubcortical temporal bilateral, en pedúnculo cerebral izquierdo y tectum mesencefálico derecho (flechas naranjas). En conjunto, dados los antecedentes de la paciente, los hallazgos sugieren origen microembólico, probablemente en el contexto de una suelta embolígena durante el episodio de disección o la cirugía aórtica.
Discusión
La enfermedad de la aorta torácica hereditaria autosómica dominante fue descrita por Guo et al. (5) en 2007 por la presencia de mutaciones heterocigotas en el gen ACTA2 que codifica la proteína alfa actina del musculo liso. La presentación clínica de la misma estaba regida por la disección de la aorta torácica asociado a iris flocculus, conducto arterioso permeable y livedo reticularis. Además, en torno al 12% de los pacientes con disección de aorta con mutación en ACTA2 presentan una valvulopatía bicuspidea (6), como el caso que presentamos.
La mutación en el gen ACTA2 condiciona el reemplazo de la arginina por la histidina en el codón 258 de la proteína ACTA2 (p. Arg258His). Existe una pequeña diferencia fisicoquímica entre la arginina y la histidina, sin embargo, el residuo de arginina se considera altamente conservado. Aunque esta variante no se encuentre en las bases de datos poblacionales, se ha informado en varias familias. Guo et al. (7) realizaron un estudio en el que analizaron 12 familias con varios miembros afectados de disección o aneurisma aórtica. Tres de estas familias que tenían mutaciones en arginina 258, presentaron ictus prematuros y 10 de 14 portadores tuvieron enfermedad aórtica. Estos ictus como parte de la enfermedad de Moyamoya, fueron resultado de la proliferación fibrocelular en la porción supraclinoidea de la arteria carótida interna. Sin embargo, revisiones criticas posteriores de las imágenes publicadas han consensuado que el fenotipo cerebrovascular de esta mutación es una entidad distinta de la enfermedad Moyamoya clásica (8). En 2015, el mismo grupo de investigación analizó 81 familias con 41 mutaciones diferentes de ACTA2. Dicho estudio concluyó que el análisis genético debe considerarse determinantemente en pacientes con antecedentes personales o familiares de aneurisma o disección aórtica torácica o variantes asociadas a la enfermedad de la aorta torácica hereditaria autosómica dominante. El diagnóstico precoz de portadores de esta mutación y el seguimiento clínico de estos pacientes con controles periódicos de la tensión arterial pueden reducir el riesgo de complicaciones y la mortalidad (9). Sin embargo, cabe destacar que el caso informado presentó una valvulopatía de inicio muy temprano. Es por ello que esta presentación debería de ser un criterio de cribado genético de la mutación ACTA2.
Por otro lado, se está estudiando la influencia de algunos factores como GDF11 en el cambio fenotípico de células musculares lisas vasculares en modelos de ratón con disección de aorta torácica. El daño arterial inducido por β-aminopropionitrilo monofumarato asociado a angiotensina II en una disección aórtica fue disminuido por la expresión excesiva de GDF11 de manera exógena por mediación del virus adenoasociado tipo 2. Esto fue posible gracias a la vía de señalización del TGF- β que propició la inhibición en la síntesis de metaloproteinasas y la remodelación de la matriz extracelular que permitieron mantener la contractibilidad de las células musculares lisas de la pared arterial (10).
Agradecimientos
Este trabajo ha sido realizado gracias a las practicas extracurriculares de verano (PEMA) de la Facultad de Medicina de la Universidad de Granada (UGR) en la convocatoria de 2021 para la plaza ofertada en el Servicio de Neurología del Hospital Universitario Clínico San Cecilio de Granada.
Referencias bibliográficas
- ↑Yang B, Norton EL, Rosati CM, Wu X, Kim KM, Khaja MS, et al. Managing patients with acute type A aortic dissection and mesenteric malperfusion syndrome: A 20-year experience. J Thorac Cardiovasc Surg. 2019;158(3):675-687.e4. https://doi.org/10.1016/j.jtcvs.2018.11.127
- ↑Houben IB, Patel HJ. Acute Type A Aortic Dissection: Managing More Than Just the Entry-Tear. Semin Thorac Cardiovasc Surg. 2019;31(1):122-128. https://doi.org/10.1053/j.semtcvs.2018.08.011
- ↑Morisaki H, Akutsu K, Ogino H, Kondo N, Yamanaka I, Tsutsumi Y, et al. Mutation of ACTA2 gene as an important cause of familial and nonfamilial nonsyndromatic thoracic aortic aneurysm and/or dissection (TAAD). Human Mutation. 2009;30(10):1406-11. https://doi.org/10.1002/humu.21081
- ↑Wang Z, Zhuang X, Chen B, Wen J, Peng F, Liu X, et al. 99-Case Study of Sporadic Aortic Dissection by Whole Exome Sequencing Indicated Novel Disease-Associated Genes and Variants in Chinese Population. Biomed Res Int. 2020:7857043. https://doi.org/10.1155/2020/7857043
- ↑Guo D-C, Pannu H, Tran-Fadulu V, Papke CL, Yu RK, Avidan N, et al. Mutations in smooth muscle α-actin (ACTA2) lead to thoracic aortic aneurysms and dissections. Nat Genet. 2007;39(12):1488-93. https://doi.org/10.1038/ng.2007.6
- ↑Martín M, Lorca R, Rozado J, Alvarez-Cabo R, Calvo J, Pascual I, et al. Bicuspid aortic valve syndrome: a multidisciplinary approach for a complex entity. J Thorac Dis. 2017;9(Suppl 6):S454-64. https://doi.org/10.21037/jtd.2017.05.11
- ↑Guo D-C, Papke CL, Tran-Fadulu V, Regalado ES, Avidan N, Johnson RJ, et al. Mutations in Smooth Muscle Alpha-Actin (ACTA2) Cause Coronary Artery Disease, Stroke, and Moyamoya Disease, Along with Thoracic Aortic Disease. Am J Hum Genet. 2009;84(5):617-27. https://doi.org/10.1016/j.ajhg.2009.04.007
- ↑Diness BR, Palmquist RN, Norling R, Hove H, Bundgaard H, Hertz JM, et al. Expanding the cerebrovascular phenotype of the p.R258H variant in ACTA2 related hereditary thoracic aortic disease (HTAD). Journal of the Neurological Sciences. 2020;415:116897. https://doi.org/10.1016/j.jns.2020.116897
- ↑Regalado ES, Guo D, Prakash S, Bensend TA, Flynn K, Estrera A, et al. Aortic Disease Presentation and Outcome Associated with ACTA2 mutations. Circ Cardiovasc Genet. 2015;8(3):457-64. https://doi.org/10.1161/CIRCGENETICS.114.000943
- ↑Ren K, Li B, Liu Z, Xia L, Zhai M, Wei X, et al. GDF11 prevents the formation of thoracic aortic dissection in mice: Promotion of contractile transition of aortic SMCs. J Cell Mol Med. 2021;25(10):4623-36. https://doi.org/10.1111/jcmm.16312
INFORMACIÓN DEL ARTÍCULO
Conflicto de intereses: Los autores de este artículo declaran no tener ningún tipo de conflicto de intereses respecto a lo expuesto en el presente trabajo.
Correspondencia: Pablo Redruello Guerrero. Avenida de la Investigación nº 11, 18016, Granada, España. E-mail: pablorg239@correo.ugr.es





