

Caso Clínico
Diagnóstico genético de hipospadia pseudovaginal perineoescrotal en paciente adolescente
Genetic diagnosis of pseudovaginal perineoscrotal hypospadias in an adolescent patient
Actual. Med. 2020; 105 : (810): 125-127 DOI: 10.15568/am.2020.810.cc03
Enviado: 15-11-2018
Revisado: 28-05-2020
Aceptado: 09-06-2020
Resumen
El déficit enzimático de 5α-reductasa tipo 2 produce una forma de pseudohermafroditismo masculino en el cual un paciente de sexo cromosómico masculino (46XY) muestra genitales ambiguos al nacer, hipospadias y desarrollo de la masculinización en la pubertad. Presentamos un caso de una paciente adolescente con genitales externos femeninos y amenorrea primaria que consulta por sospecha de acromegalia. Se realiza un estudio hormonal, de imagen y genético que revela un déficit de 5-alfa-reductasa tipo 2, con la presencia de la variante c.271T>G (p.Tyr91Asp) en homocigosis. La tardanza en el diagnóstico supuso un retraso en la instauración de un régimen terapéutico adecuado, provocando una androgenización excesiva de la paciente con la consecuente afectación psicológica.
Palabras clave: 5-alfa-reductasa; Hermafroditismo; Hipospadia pseudovaginal perineoescrotal; Diagnóstico genético.
Abstract
5-alpha-reductase enzymatic deficit produces a form of pseudohermaphroditism in which a chromosomic male (46XY) shows ambiguous genitalia at birth, hypospadias and masculinization at puberty. We present a clinical case of a teenage patient with female external genitalia and primary amenorrhoea who was derived from her family doctor on suspicion of acromegaly. Hormonal, imaging and genetic research was conducted and it revealed a 5-alpha-reductase type 2, presenting the variant c.271T>G (p.Tyr91Asp) in homozygosity. A diagnosis delay suppossed a lag in appropiate therapeutic regime instauration, thus leading to an excessive androgenization of the patient with the corresponding psychological distress.
Keywords: 5-alpha-reductase; Hermaphroditism; Pseudovaginal perineoscrotal Hypospadias; Genetic diagnosis.
Leer Artículo Completo
INTRODUCCIÓN
Las esteroide 5α-reductasas son un conjunto de enzimas encargadas de la reducción del doble enlace entre los carbonos 4 y 5 de los 3-oxo-Δ4,5 C19/C21 esteroides. Existen tres isoenzimas, productos de los genes SRD5A1, SRD5A2 y SRD5A3, que en humanos tienen diferentes niveles de expresión según tejido y edad del individuo. Así, durante el desarrollo fetal, la isoenzima 1 se expresa fundamentalmente en la piel y la 2 en la próstata fetal, mientras que después del nacimiento la expresión incluye órganos como el hígado, la piel y la próstata para la isoenzima 1, mientras que la isoenzima 2 lo hace en próstata, glándulas seminales, epidídimo, hígado, piel y folículo piloso. La isoenzima 3 se expresa en todos estos tejidos solo durante la edad adulta(1).
Los sustratos sobre los que actúan las 5α-reductasas incluyen diversos esteroides. Sin embargo, su función mejor conocida es la transformación de testosterona en dihidrotestosterona (DHT), un metabolito que se une con mayor afinidad que la propia testosterona al receptor de andrógenos. La DHT se encarga de inducir el desarrollo sexual masculino durante el periodo fetal, y más tarde durante la pubertad(2).
El gen que codifica la isoenzima 2 (SRD5A2) está localizado en el cromosoma 2p23.1, consta de 5 exones y 4 intrones. Cualquier mutación que produzca una pérdida parcial o total de su actividad, puede afectar al desarrollo sexual normal del individuo. Se han descrito más de 100 mutaciones(3), repartidas a lo largo de todo el gen, y sin una clara agregación geográfica, salvo en casos de comunidades cerradas con alta endogamia(4). Los casos reportados muestran una ligera mayoría de homocigotos frente a heterocigotos compuestos, y no parece haber una clara asociación genotipo-fenotipo. Las mutaciones provocan desde una reducción de la actividad enzimática hasta la pérdida total de la misma, lo que explica la variedad de presentación clínica del síndrome. Fenotípicamente, los signos más reportados son clitoromegalia y micropene, diferentes grados de hipospadias, y criptoorquidismo unilateral o bilateral. Todos los casos presentan una virilización en la pubertad que puede llevar al cambio sexual, de mujer a hombre. Esta virilización se produce debido a la actividad de la isoenzima 5α-reductasa tipo 1, que se mantiene en los tejidos donde se expresa, provocando la conversión de testosterona en DHT en los mismos(1).
Dentro del espectro de desórdenes del desarrollo sexual causados por mutaciones en este gen, la hipospadia perineoscrotal pseudovaginal (PPSH), también llamado pseudohermafroditismo familiar masculino incompleto tipo 2 (OMIM #264600), es una forma de pseudohermafroditismo masculino en el que individuos cromosómicamente de sexo masculino, que presentan genitales ambiguos al nacer, incluyendo hipospadias perineales con un saco pseudovaginal ciego, desarrollan masculinización en la pubertad.
Descripción del caso clínico
Paciente de sexo femenino derivada a endocrinología desde atención primaria a los 14 años de edad por sospecha de acromegalia, sin antecedentes personales ni familiares de interés, salvo hermano mayor que presenta ginecomastia sin otras alteraciones. En la exploración presentó un peso de 74.6 kg, talla 178 cm e IMC 23.54 kg/m², sin menarquia y sin presentar otros signos de disfunción hormonal.
Se realizó radiografía de mano izquierda sin anomalías y se siguió su evolución durante un periodo de un año con perfil hormonal en el que destacó testosterona elevada (hasta 5.19 ng/mL, intervalo de referencia 0.1 a 0.8 ng/mL) y tras comprobar la ausencia de rasgos físicos característicos de acromegalia, se descartó dicha patología.
La paciente fue seguida durante un año por el servicio de endocrinología, produciéndose en dicho intervalo de tiempo una ganancia de peso de 10 Kg. En este momento se plantea el diagnóstico diferencial entre disgenesia gonadal y síndrome de resistencia parcial androgénica, derivándose a revisión por ginecología. A la exploración física se encontraron mamas no desarrolladas, genitales externos con vello púbico e hipertrofia notable del órgano eréctil. En ingles se palparon nódulos móviles de 2 cms. Se le realizó una resonancia magnética nuclear (RMN) de pelvis con contraste donde no se visualizó ni útero ni ovarios, evidenciándose un residuo vaginal de 1cm, con imagen sugerente de senos cavernosos pequeños, pene hipoplásico y testículos no descendidos (figura 1), localizados a nivel de tejido celular subcutáneo inguinal bilateral (criptorquidia bilateral). Se efectuó un análisis citogenético encontrándose una fórmula cromosómica 46XY. Ante este resultado se confirma la presencia de un hermafroditismo y la paciente es derivada a consulta de atención a las personas transexuales, desde donde se solicita estudio genético de déficit de 5-alfa-reductasa de tipo 2, encontrándose la variante c.271T>G (p.Tyr91Asp) en homozigosis del gen de SRD5A2. A continuación se programó orquiectomía inguinal bilateral y se inició tratamiento hormonal sustitutorio con estradiol y triptorelina (análogo GnRH).
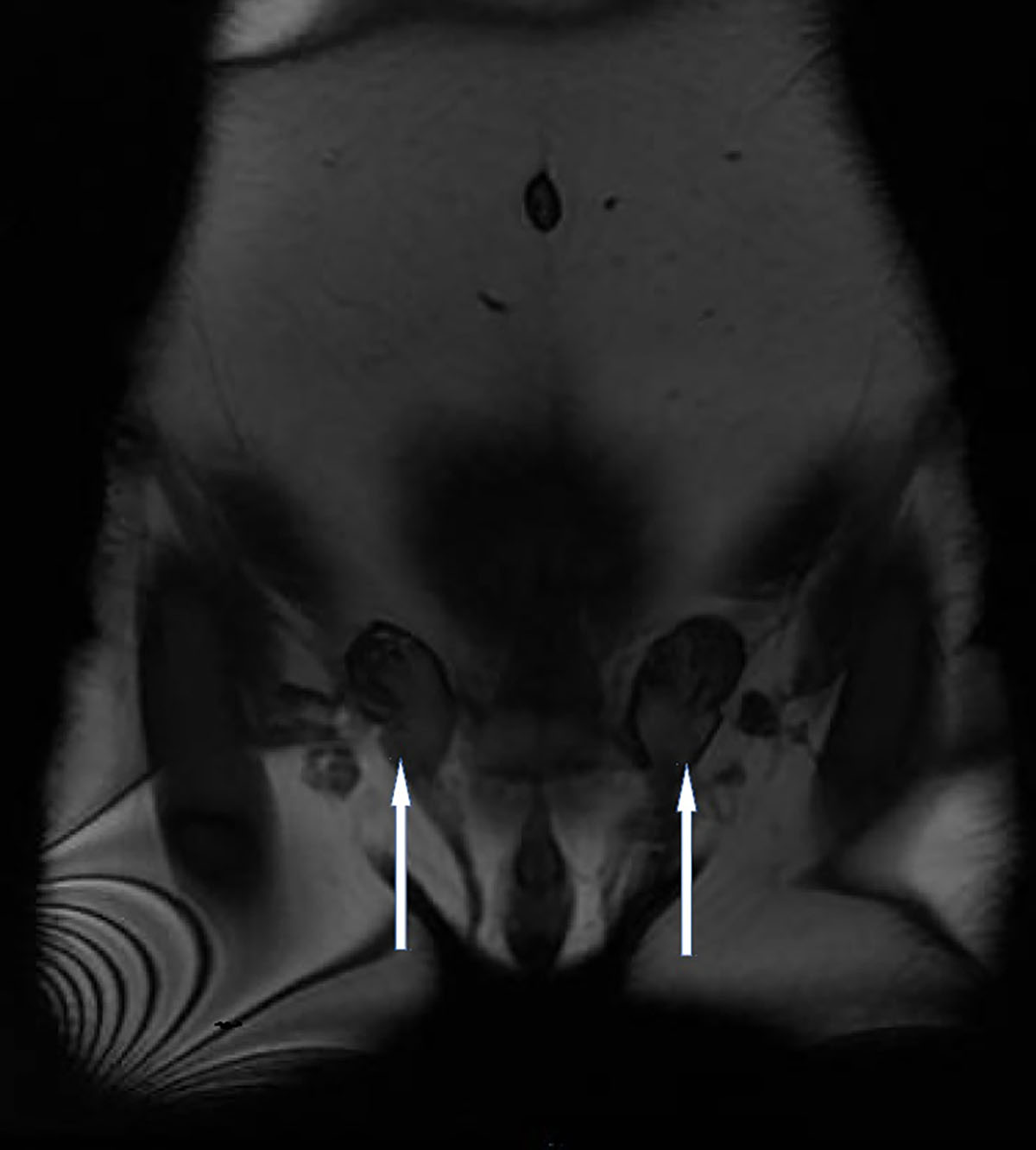
Figura 1. Imagen de resonancia magnética nuclear de pelvis en la que se observa criptorquidismo.
Discusión
La variante genética encontrada en la paciente, c.271T>G (p.Tyr91Asp), se halla en un locus que tiene tres posibles alelos (A, G y C). El alelo A o ancestral es el encontrado en la secuencia de referencia y las variantes alélicas G y C corresponden a las mutaciones c.271T>C (p.Tyr91His) y c.271T>G (pTyr91Asp). Ambas variantes están únicamente reportadas como patogénicas una sola vez en Clinvar en el 2017 y 2010, respectivamente, por el Clinical Molecular Genetics Laboratory (Johns Hopkins All Children’s Hospital).
Establecer un diagnóstico correcto es esencial para poder instaurar un manejo correcto de los pacientes. La PPSH representa un diagnóstico diferencial con el síndrome de feminización testicular incompleta, también conocido como pseudohermafroditismo masculino familiar incompleto de tipo 1 o síndrome de Reifenstein. La PPSH recuerda a la forma más severa de este síndrome, pero difiere en la ausencia de desarrollo mamario, así como en su modo de herencia y riesgo de recurrencia. La PPSH tiene un modo de herencia autosómico recesivo y afecta solo a individuos de sexo masculino, mientras que el síndrome de Reifenstein es un trastorno recesivo ligado al cromosoma X(5,6).
Para el diagnóstico, un valor elevado de la relación T/DHT tras estimulación con gonadotropina coriónica humana (hCG), con un punto de corte de 18-20, es sugestivo de déficit de SRD5A2. No obstante un estudio del perfil de andrógenos en orina es mucho más preciso. El diagnóstico molecular del gen SRD5A2 es considerado no obstante como el test diagnóstico más efectivo.
El tratamiento dependerá de la identidad sexual del paciente, idealmente se recomienda hacer una reasignación del sexo antes de los 27 meses de edad para evitar posibles conflictos de identidad. En los pacientes masculinos la testosterona no suele ser necesaria ya que la mayoría de ellos han mantenido la función testicular durante la pubertad. Sin embargo inyecciones de testosterona intramuscular o gel de dihidrotestosterona pueden mejorar el vello corporal y el tamaño del pene. En general se recomienda el empleo del gel de dihidrotestosterona al ser más activo y conseguir efectos más rápidos, ademas de tener la ventaja de no ser un andrógeno aromatizable.
Para los pacientes criados como mujeres el objetivo de la terapia hormonal es conseguir el desarrollo de las características sexuales femeninas. Se deben dar bajas dosis de estrógenos (0,07-0,3 mg de estrógeno conjugado) al inicio de la edad puberal y mantener hasta el desarrollo mamario, y posteriormente mantener dosis más altas de manera continua (0,625-1,25 mg/día). El tratamiento quirúrgico consistirá en conseguir unos genitales externos de acuerdo con la identidad sexual y eliminar las estructuras incompatibles con esta, en caso de adoptar un género femenino.
En el caso presentado, tras evaluar psicológicamente a la paciente y teniendo en cuenta sus preferencias se programa cirugía correctora y se inicia tratamiento hormonal sustitutorio con estradiol y triptorelina (análogo de la hormona liberadora de gonadotropina, GnRH). Actualmente la paciente continua con el tratamiento farmacológico prescrito y a la espera de la intervención quirúrgica.
REFERENCIAS BIBLIOGRÁFICAS
- ↑ Azzouni F, Godoy A, Li Y, Mohler J. The 5 alpha-reductase isozyme family: A review of basic biology and their role in human diseases. Adv Urol. 2012;2012.
- ↑ Okeigwe I, Kuohung W. 5-Alpha reductase deficiency: A 40-year retrospective review. Curr Opin Endocrinol Diabetes Obes. 2014;21(6):483–7.
- ↑ Avendaño A, Paradisi I, Cammarata-Scalisi F, Callea M. 5-α-Reductase type 2 deficiency: is there a genotype-phenotype correlation? A review. Hormones. 2018;17(2):197–204.
- ↑Alswailem MM, Alzahrani OS, Alghofaili L, Qasem E, Almohanaa M, Alsagheir A, et al. Molecular genetics and phenotype/genotype correlation of 5-α reductase deficiency in a highly consanguineous population. Endocrine [Internet]. 2019;63(2):361–8. Available from: http://dx.doi.org/10.1007/s12020-018-1767-1
- ↑ Lee PA, Houk CP, Ahmed SF, Hughes IA, Achermann J, Ahmed F, et al. Consensus statement on management of intersex disorders. Pediatrics. 2006;118(2).
- ↑Turan V, Yeniel Ö, Ergenoǧlu M, Terek C, Ulukuş M. Incomplete androgen insensitivity (Reifenstein syndrome) – a case report. J Turkish Ger Gynecol Assoc. 2010;11(2):110–2.
INFORMACIÓN DEL ARTÍCULO
Los autores declaran no tener conflicto de intereses.
Autor para la correspondencia: Alfredo Pérez-Alija Fernández. UGC Laboratorios, Hospital la Inmaculada. Av./ Dtra. Ana Parra s/n · 04600 Huércal-Overa, Almería. E-mail: alfredo.perezalija.sspa@juntadeandalucia.es





