

Caso Clínico
Anomalía de Rieger y síndrome de Axenfeld-Rieger: rara disgenesia del segmento anterior
Rieger anomaly and Axenfeld-Rieger syndrome: a rare anterior segment dysgenesis
Actual Med. 2021; 106(813): 218-221 DOI: 10.15568/am.2021.813.cc06
Recibido: 20/02/2021
Revisado: 01/04/2021
Aceptado: 20/05/2021
Resumen
El Síndrome de Axenfeld Rieger es una disgenesia del segmento anterior caracterizada por embriotoxon posterior y disgenesia de la cámara anterior. Las anormalidades en el desarrollo del ángulo causan una mayor resistencia al flujo de salida del humor acuoso, pudiendo desarrollarse hipertensión en casi el 50% de los casos. El Síndrome de Axenfeld Rieger es una enfermedad autosómica dominante, con una prevalencia de 50.000-100.000 recién nacidos por año. Los principales genes alterados son el FOXC1 y el PITX2, ocurriendo en el 40% de los casos. El Síndrome de Axenfeld Rieger puede asociarse con manifestaciones sistémicas como anomalías dentales (hipodoncia y microdoncia), anomalías faciales (hipoplasia maxilar, telecanto e hipertelorismo), piel paraumbilical redundante, hipospadia, alteraciones auditivas y cardíacas. Dentro de este síndrome, distinguimos diferentes fenotipos, siendo la Anomalía de Rieger la de menor afectación sistémica. Presentamos tres casos diferentes de la enfermedad y su manejo.
Palabras clave: Síndrome de Axenfeld Rieger; Anomalía de Rieger; Disgenesia del segmento anterior; Glaucoma; Segmento anterior.
Abstract
Axenfeld Rieger Syndrome is anterior segment dysgenesis characterized by posterior embryotoxon and dysgenesis of the anterior chamber. Developmental abnormalities of the anterior angle cause increased resistance to outflow. Ocular hypertension is a complication in almost 50% of the cases. It is an autosomal dominant disease and its prevalence is between 50,000 and 100,000 newborns per year. The main associated affected genes are FOXC1 and PITX2, occurring in 40% of the cases. Axenfeld Rieger Syndrome can be associated with systemic manifestations such as dental anomalies (hypodontia and microdontia), facial anomalies (maxillary hypoplasia, telecanthus and hypertelorism), redundant paraumbilical skin, hypospadia, auditory and cardiac alterations. Within this syndrome, we distinguish different phenotypes, being the Rieger anomaly the one with least systemic affectation. We present three cases in different stages of the disease, which allow us to understand the development and management of this disorder.
Keywords: Axenfeld Rieger syndrome; Rieger anomaly; Anterior segment dysgenesis; Glaucoma; Anterior segment.
Leer Artículo Completo
INTRODUCCIÓN
El Síndrome de Axenfeld-Rieger (SAR) fue descrito por el oftalmólogo alemán Axenfeld en 1920. Describió el caso de un paciente que mostró un avance de la línea de Schwalbe (denominándolo embriotoxón posterior, EP) y fibras del iris dirigidas hacia esta línea. En 1934, Rieger detalló una serie de casos con estas anomalías, alteraciones en el iris y de la pupila. (1,2,3,4)
El SAR consiste en una disgenesia del segmento anterior caracterizada por EP y disgenesia de la cámara anterior. Las anomalías en el desarrollo del ángulo causan una mayor resistencia a la salida del humor acuoso, desarrollando hipertensión ocular en el 50% de los casos. En el pasado, esta enfermedad se definió bajo tres subcategorías: 1) Anomalía de Axenfeld: EP con inserción de fibras de iris periférico; 2) Anomalía de Rieger (AR): EP con alteraciones del iris y pupila, pudiendo variar desde una ligera hipoplasia del estroma iridiano hasta una afectación más extensa con policoria, corectopía y ectropión uveal; 3) SAR: AR con manifestaciones sistémicas tales como anomalías dentales (hipodoncia y microdoncia), anomalías faciales (hipoplasia maxilar, telecantus e hipertelorismo), piel paraumbilical redundante, hipopasdias, alteraciones auditivas y/o cardíacas. (1–5)
El objetivo de nuestro trabajo es presentar tres casos; cada uno presenta un espectro distinto dentro de dicho síndrome. Realmente la recolección de los estos distintos espectros nos permite conocer la distinta presentación de la enfermedad, así como su manejo. En nuestros casos se realizó tanto tratamiento conservador, medicamentoso e incluso invasivo (quirúrgico).
CASO CLÍNICO UNO
Paciente de 13 años acudió a la consulta por diagnóstico de AR e hipertensión ocular en el ojo izquierdo (OI). La mejor agudeza visual corregida de la paciente (MAVC) fue de 0,8 Snellen en el ojo derecho (OD) y 0,4 en el OI. La presión intraocular (PIO) del OI fue de 34 mm Hg tratada con bimatoprost una vez al día y 14 mm Hg en OD sin tratamiento. El examen con lámpara de hendidura del OD fue normal. En OI se observó córnea clara, policoria y corectopía temporal (Figura 1. A, B y C). Debido al aumento de la PIO, se prescribió dorzolamida y timolol dos veces al día. La funduscopia fue normal en ambos ojos (AO) y no hubo manifestaciones sistémicas.
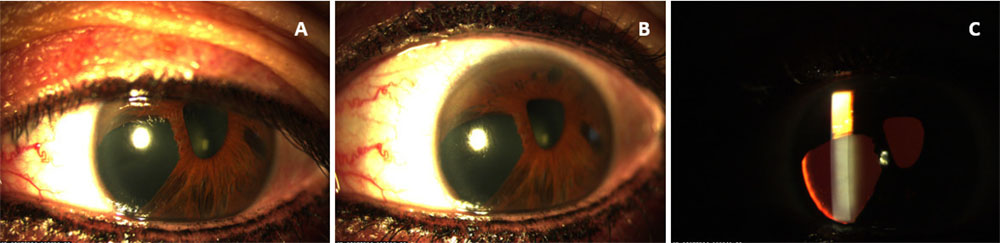
Figura 1. A y B. Fotografía con lámpara de hendidura del ojo izquierdo con conjuntiva normal, córnea clara y policoria. C. Reflejo rojo mediante transiluminación que permite ver la policoria.
CASO CLÍNICO DOS
Paciente de 4 años acudió a consulta por tener unas manchas oscuras en AO desde el nacimiento. La historia familiar presentaba antecedentes de disgenesia iridiana. MAVC fue de 1.0 (AO) y la PIO de 16 mmHg (AO).
En AO se observaron córneas claras, EP, corectopía, atrofia del iris estromal y adherencias iridocorneales (Figuras 2. A, B y C).
Mediante gonioscopia se vió una prominente línea de Schwalbe con puentes de tejido iridiano en AO. La funduscopia fue normal.
Sistémicamente, el paciente presentaba un leve dismorfismo craneofacial con hipoplasia maxilar, aplanamiento de la cara, frente prominente, puente nasal ancho y plano, microdoncia y retraso del crecimiento (Figuras 2. D, E y F).
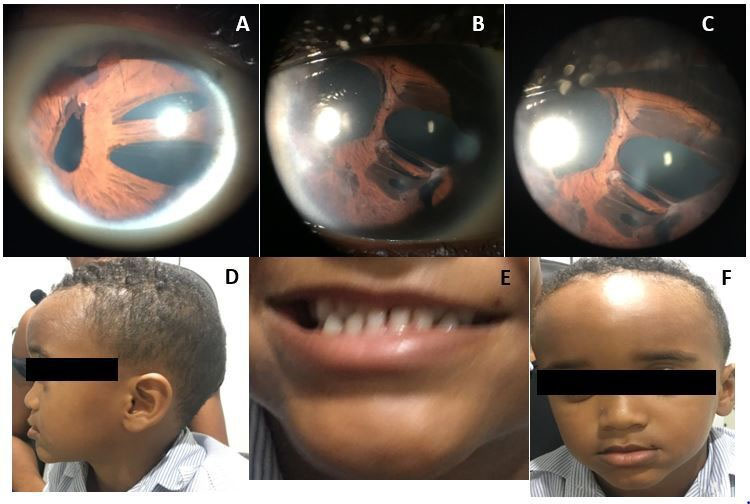
Figura 2. A. Fotografía con lámpara de hendidura (LH) del ojo derecho: córnea clara y corectopía. B y C. LH del ojo izquierdo: córnea clara, corectopía con atrofia del iris. D, E y F. Dismorfismo craneofacial leve como hipoplasia maxilar, aplanamiento de la cara, frente prominente, puente nasal amplio y plano y microdoncia.
CASO CLÍNICO TRES
Paciente de 14 años acude a consulta, en seguimiento por AR y glaucoma secundario, sin antecedente familiares relevantes. El paciente había sido sometido a una trabeculectomía (dos veces en AO) y una válvula de Ahmed en OI. MAVC era de 0.4 (AO). La PIO fue de 10 mm Hg y 22 mm Hg sin tratamiento en OD y OI, respectivamente.
El examen con lámpara de hendidura reveló policoria, corectopía bilateral y válvula de Ahmed en el OI (Figura 3. A y B). La funduscopia fue normal excepto por un disco óptico pálido y excavado (0.8) en OI.
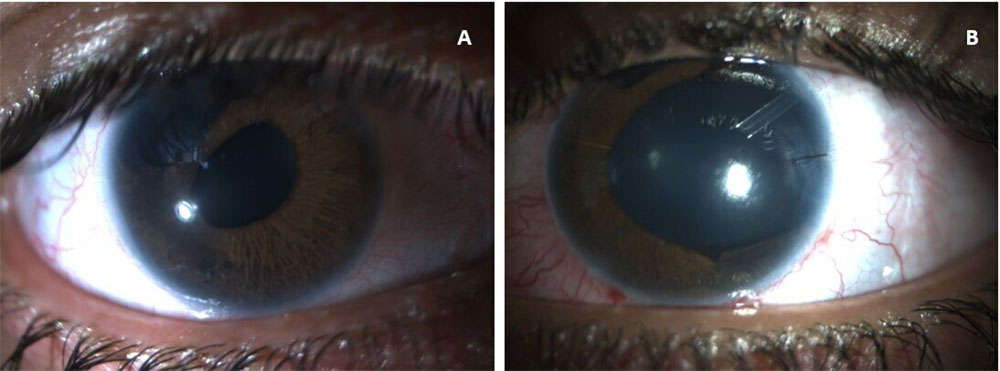
Figura 3. A. Fotografía con lámpara de hendidura (LH) del ojo derecho: córnea clara y anomalías iridiales. B. LH del ojo izquierdo: con corectopía y válvula de Ahmed.
La gonioscopia mostró un ángulo pigmentado con procesos irido-trabeculares.
No hubó manifestaciones sistémicas.
DISCUSIÓN
El SAR es una enfermedad autosómica dominante (EAD) que afecta a las estructuras derivadas del mesénquimal periocular (MP). Se cree que esta patología se produce debido a una alteración durante la embriogénesis entre el primer y el cuarto mes.(1) La causa podría ser una migración anormal de las células derivadas de la cresta neural durante las primeras etapas del desarrollo, debido a una mutación en los factores de transcripción, los cuales regulan estos procesos. En las fases tardías, el endotelio primordial debe sufrir un proceso de apoptosis que no se produce en esos casos, causando la aparición del subsiguiente EP, inserción anormal del iris y los cambios de la pupila. Todas estas alteraciones aumentan el riesgo de glaucoma. Dado que las células de la cresta neural también están presentes en la formación de otros tejidos extraoculares, las alteraciones extraoculares también pueden ocurrir (deterioro de la audición, alteraciones cardiacas, etc). (2,3,5)
En cuanto a la genética, el SAR es una EAD en el 75% de los casos, aunque puede ser esporádica. (3,6) Los principales genes afectados (40%) son el gen Forkhead-Box C1 (FOXC1) y el gen pituitario Homeobox 2 (PITX2), ambos genes codificadores de la transcripción. (1,3) El FOXC1 se expresa en gran medida en el MP y su función es asegurar la estabilidad de la célula (6); mientras que el PITX2 regula la formación de los tejidos oculares y no oculares (dientes, corazón y riñones). (1) A pesar de todas las investigaciones realizadas, los defectos genéticos asociados al 60% de esta patología siguen siendo desconocidos. (1,3) Selfi et al. (1) demostraron que el tratamiento médico no consigue reducir la PIO en los pacientes con SAR asociados a las mutaciones de FOXC1 o PITX2. En estos casos, los tratamientos con betabloqueantes tópicos están contraindicados ya que la SAR puede relacionarse con problemas cardíacos. (1)
En los pacientes pediátricos, la trabeculotomía y la trabeculectomía son eficaces para el glaucoma de inicio temprano asociado con SAR, mientras que la goniotomía tiene resultados deficientes. (2) Sin embargo, al estar nuestros pacientes en un rango a medio camino entre la edad pediátrica tardía y la adulta, se decidió comenzar con tratamiento médico, ofreciendo la trabeculectomía como segunda opción. Nuestro tercer paciente mostró una gran respuesta a la trabeculectomía y a la implantación de la válvula de Ahmed. Otras series de casos (2,7,8) también han mostrado buenos resultados con el mismo tratamiento, lo que apoya nuestra elección quirúrgica, desaconsejando el uso de la goniotomía debido a que pueden provocar un desprendimiento de la membrana de Descemet al tratar de cortar el tejido anormal del ángulo camerular. (2)
Strungaru et al. (9), en un estudio retrospectivo (n=126), informaron que el glaucoma asociado a SAR no responde bien al tratamiento médico convencional. Doucette et al. (10) asociaron la falta de respuesta a los análogos de la prostaglandina debido a la mutación FOXC1, un gen que regula la vía de señalización de la prostaglandina. Por lo tanto, las intervenciones quirúrgicas parecen ser actualmente el mejor tratamiento para el glaucoma en los pacientes con SAR. En nuestros casos, todas estas conclusiones fueron sopesadas, eligiéndose la opción médica o quirúrgica según el espectro y el rango de edad. Posiblemente, un mayor estudio de las mutaciones y las vías de señalización permita un mejor control del glaucoma en el futuro. (3)
En resumen, los autores creemos que es necesario realizar estudios sobre el tratamiento del glaucoma en pacientes con SAR para comprender mejor esta enfermedad y definir protocolos de tratamiento que puedan mejorar el pronóstico de estos pacientes. En nuestro caso, presentamos tres pacientes distintos, con tres abordajes diferentes que permiten un mejor entendimiento de esta patología.
REFERENCIAS BIBLIOGRÁFICAS
- ↑ Seifi M, Walter MA. Axenfeld-Rieger syndrome. Clin Genet. 2018; 93(6): 1123–30. DOI: 10.1111/cge.13148
- ↑ Mandal AK, Pehere N. Early-onset glaucoma in Axenfeld–Rieger anomaly: long-term surgical results and visual outcome. Eye. 2016; 30(7): 936–42. DOI: 10.1038/eye.2016.66
- ↑ Chrystal PW, Walter MA. Aniridia and Axenfeld-Rieger Syndrome: Clinical presentations, molecular genetics and current/emerging therapies. Exp Eye Res. 2019. 189: 107815. DOI: 10.1016/j.exer.2019.107815
- ↑ Song W, Hu X. The rare Axenfeld-Rieger syndrome with systemic anomalies: A case report and brief review of literature. Med. 2017; 96(33): e7791. DOI: 10.1097/MD.0000000000007791
- ↑ Craig Dunbar A, McIntyre GT, Laverick S, Stevenson B. Axenfeld-Rieger syndrome: A case report. J Orthod. 2015; 42(4): 324-30. DOI: 10.1179/1465313315Y.0000000017
- ↑ Berry FB, Lines MA, Oas JM, Footz T, Underhill DA, Gage PJ, et al. Functional interactions between FOXC1 and PITX2 underlie the sensitivity to FOXC1 gene dose in Axenfeld-Rieger syndrome and anterior segment dysgenesis. Hum Mol Genet. 2006; 15(6): 905–19. DOI: 10.1093/hmg/ddl008
- ↑ Beck AD, Wilson WR, Lynch MG, Lynn MJ, Noe R. Trabeculectomy with adjunctive mitomycin C in pediatric glaucoma. Am J Ophthalmol. 1998; 126(5): 648–57. DOI: 10.1016/s0002-9394(98)00227-x
- ↑ Sidoti PA, Belmonte SJ, Liebmann JM, Ritch R. Trabeculectomy with mitomycin-C in the treatment of pediatric glaucomas. Ophthalmology. 2000; 107(3): 422–9. DOI: 10.1016/s0161-6420(99)00130-x
- ↑ Strungaru MH, Dinu I, Walter MA. Genotype-phenotype correlations in Axenfeld-Rieger malformation and glaucoma patients with FOXC1 and PITX2 mutations. Invest Ophthalmol Vis Sci. 2007; 48(1): 228–37. DOI: 10.1167/iovs.06-0472
- ↑ Doucette LP, Footz T, Walter MA. FOXC1 regulates expression of prostaglandin receptors leading to an attenuated response to latanoprost. Invest Ophthalmol Vis Sci. 2018; 59(6): 2548–54. DOI: 10.1167/iovs.17-23223
INFORMACIÓN DEL ARTÍCULO
Conflicto de intereses: Los autores/as de este artículo declaran no tener ningún tipo de conflicto de intereses respecto a lo expuesto en el presente trabajo.
Autor para la correspondencia: Javier Lacorzana. Hospital Virgen de las Nieves. Av. de las Fuerzas Armadas, 2 · 18014 Granada, España. E-mail: javilacor@gmail.com





