

Revisión
Secuenciación del genoma del Sars-CoV-2 en Andalucía, metodología y estudio de las variantes
SARS-CoV-2 genome sequencing in Andalusia, methodology and study of variants
Actual Med. 2021; 106(814): 291-300 DOI: 10.15568/am.2021.814.rev03
Recibido: 19/09/2021
Revisado: 07/12/2021
Aceptado: 06/02/2022
RESUMEN
La incorporación de las técnicas de secuenciación genómica mediante secuenciación de nueva generación ha revolucionado la microbiología clínica, innovando y mejorando el diagnóstico clínico de las enfermedades infecciosas. Hoy en día, la secuenciación de genoma completo en enfermedades infecciosas tiene multitud de aplicaciones en virología, bacteriología, resistencia antibiótica, epidemiología y salud pública. Con la aparición del SARS-CoV-2, se ha visto subrayada la importancia del análisis y estudio de las secuencias genéticas. Desde la identificación inicial del SARS-CoV-2, hasta la fecha, se han compartido, a nivel mundial, más de 414.575 secuencias genómicas completas a través de bases de datos de acceso público. La capacidad de monitorizar la evolución viral casi en tiempo real tiene un impacto directo en la respuesta de salud pública a la pandemia de COVID-19.
En este trabajo se presenta la importancia de la secuenciación genómica en microbiología, enfermedades infecciosas, epidemiología y salud pública, y se describe cómo se ha implementado la secuenciación de SARS-CoV-2 en Andalucía, y cuales son los principales resultados hasta la fecha
Palabras clave: Secuenciación genómica; SARS-CoV-2; Variantes; Andalucía.
ABSTRACT
The incorporation of genomic sequencing techniques through next-generation sequencing has revolutionized clinical microbiology, innovating and improving the clinical diagnosis of infectious diseases. Today, whole genome sequencing in infectious diseases has many applications in virology, bacteriology, antibiotic resistance, epidemiology, and public health. With the appearance of SARS-CoV-2, the importance of the analysis and study of genetic sequences has been underlined. Since the initial identification of SARS-CoV-2, to date, more than 414,575 complete genomic sequences have been shared worldwide through public access databases. The ability to monitor viral evolution in near real time has a direct impact on the public health response to the COVID-19 pandemic.
This paper presents the importance of genomic sequencing in microbiology, infectious diseases, epidemiology and public health, and describes how SARS-CoV-2 sequencing has been implemented in Andalusia, and what the main results are to date.
Keywords: Genomic sequencing; SARS-CoV-2; Variants; Andalusia
Leer Artículo Completo
SÍNTESIS DE LA REVISIÓN
Secuenciación genómica: bases metodológicas
El genoma constituye el material genético propio de un organismo, el cual viene determinado por las bases nucleotídicas que se unen entre sí y conforman su ADN. El proceso de secuenciación permite conocer el orden preciso de las bases nitrogenadas de una cadena de ADN, determinando su secuencia nucleotídica y el posterior análisis de la misma.
En el ámbito de la Microbiología Clínica la caracterización genética del microorganismo en cuestión nos permite identificar al patógeno involucrado en la infección, convirtiéndose en una herramienta más en el diagnóstico microbiológico.
En la actualidad, existen diversas técnicas de secuenciación (1, 2, 3):
- Secuenciación de 1ª Generación (Sanger):
Se basa en la síntesis enzimática de la cadena complementaria a aquella que se quiere secuenciar, mediante la adición de dideoxinucleótidos marcados. Estos actúan como terminadores de la cadena interrumpiendo la síntesis y se genera un fragmento de un tamaño determinado condicionado por el nucleótido incorporado; tras ser separados en un gel de electroforesis, y ser leídos posteriormente, permite la reconstrucción de la secuencia molde que queríamos conocer.
- Secuenciación de 2º Generación (Next Generation Sequencing o secuenciación de nueva generación, NGS):
Esta metodología permite un mayor rendimiento a menor coste, ya que la NGS consiste en generar millones de fragmentos de ADN en un único proceso de secuenciación. Estos fragmentos se asignan a cada una de las muestras analizadas gracias a la preparación previa de una librería que son inmovilizadas sobre una superficie.
Existen diferentes tecnologías que nos permiten la realización de NGS, pero todas tienen en común la preparación previa de la librería, inmovilización en una superficie y amplificación de esta, secuenciación y captación de la señal.
– Preparación de librería: consiste en fragmentar y marcar, empleando unos adaptadores únicos, a las diferentes muestras de ADN que quieren conocerse.
– Fijación a la superficie y amplificación.
– Reacción de Secuenciación, existen diferentes estrategias, pero todas ellas nos permiten la detección de las bases incorporadas. La secuenciación por síntesis, empleada por Ilumina, emplea nucleótidos marcados con fluorocromos, estos se unen por complementariedad de bases, cuando están unidos se excita el fluorocromo y esta señal es captada y traducida a nucleótido. La secuenciación mediante ligación, utilizada por Ion Torrent, emplea una mezcla de sondas marcadas con fluoroforos que se unen mediante ligasas.
- Secuenciación de 3º Generación (De Molécula Única):
El objetivo de esta tercera generación es la secuenciación de moléculas únicas en tiempo real, sin necesidad de fragmentar el ADN y sin que se requiera la amplificación clonal de las moléculas individuales a secuenciar. Esta tecnología es la que se emplea en los secuenciadores MiniON (4); se basa en la secuenciación de moléculas únicas mediante la detección electrónica a su paso por nanoporos, y que consiste en asociar a este nanoporo una enzima que lo que hace es obligar al ADN a pasar por él, base a base, de tal manera que el campo eléctrico que hay en el mismo se ve distorsionado de una forma concreta por cada nucleótido, con lo cual va leyendo en tiempo real cada una de estas incorporaciones cambiando la magnitud de la corriente de forma especifica para cada base permitiendo así la lectura de la hebra.
Aplicaciones de la secuenciación genómica en EEII
La incorporación de las técnicas de secuenciación genómica mediante secuenciación de nueva generación ha revolucionado la microbiología clínica, innovando y mejorando el diagnóstico clínico de las enfermedades infecciosas. Además de identificar patógenos con mayor rapidez y precisión que los métodos tradicionales, la secuenciación genómica puede proporcionar nuevos conocimientos sobre la transmisión de enfermedades, la virulencia y la resistencia a los antimicrobianos (5, 6, 7).
Hoy en día, la secuenciación de genoma completo en enfermedades infecciosas tiene multitud de aplicaciones en función del campo (8,9):
- Aplicaciones en virología:
– Identificación de nuevos agentes infecciosos virales.
– Pruebas de mutaciones de resistencia a fármacos antivirales y antirretrovirales, en el campo del virus de la inmunodeficiencia humana (VIH), virus de la hepatitis C (VHC), de la hepatitis B (VHB), Citomegalovirus (CMV) o virus de la Influenza, entre otros (10,11).
- Aplicaciones en bacteriología y resistencia antibiótica:
– Identificación y caracterización rápida de microorganismos, proporcionando información sobre la relación de las cepas, de dónde provienen y cómo han evolucionado.
– Identificación de factores clave de virulencia: características específicas que ayudan al microorganismo a causar la infección.
– Perfiles de resistencia a los antibióticos: en comparación con los métodos de cultivo tradicionales, la secuenciación genómica puede determinar a qué antibióticos los microorganismos son resistentes mucho más rápidamente y ofrecer un perfil de resistencia completo (12). Esto es especialmente interesante en microorganismos de crecimiento lento como Mycobacterium tuberculosis (13) o microorganismos no cultivables.
– Estudio de elementos genéticos móviles: integrones, transposones, plásmidos y bacteriófagos. Además del genoma completo, los elementos genéticos móviles constituyen un papel importante en la resistencia antibiótica y factores de virulencia.
- Aplicaciones en epidemiología y salud pública
– Detección, mapeo y análisis de brotes. Mediante la secuenciación genómica podemos averiguar si los aislamientos de un brote están relacionados entre sí, podemos realizar estudios filogenéticos y tipificación de aislamientos, siendo una herramienta fundamental para la epidemiología y el control de brotes (14).
– Intervenciones no farmacológicas para el control de la enfermedad. El análisis de datos genéticos con los métodos filodinámicos permite hacer inferencias sobre las características de los individuos involucrados en la transmisión de la infección y sobre cómo los patrones de contacto y la dinámica de los comportamientos de riesgo afectan el flujo de transmisión a través de una población, lo que permite adaptar la aplicación de intervenciones poblacionales y adecuar las estrategias de control (15).
Además de todo lo anterior, la tecnología usada para la secuenciación genómica ofrece ventajas en campos desafiantes. Un ejemplo lo observamos durante el brote de Ébola de 2015, en el que un equipo de investigación del Reino Unido pudo llevar un laboratorio de secuenciación de nanoporos transportado en un equipaje estándar, con el que pudieron secuenciar más de 140 genomas del virus del Ébola in situ; los datos se transmitieron a la nube para su análisis y los resultados se devolvieron al día siguiente. A pesar de los desafíos logísticos, incluida la energía eléctrica y un servicio de internet deficiente, el equipo proporcionó una información muy importante sobre la transmisión del virus, dando respuesta a la epidemia sin tener que exportar muestras del país (16).
Necesidad de la secuenciación genómica en el COVID 19
El estudio de las secuencias genómicas ha adquirido un papel fundamental en la detección y en el manejo de los brotes de enfermedades infecciosas. Con la aparición del SARS-CoV-2, se ha visto subrayada la importancia del análisis y estudio de las secuencias genéticas.
A medida que el virus se fue diseminando por el mundo, gracias a la secuenciación genómica fue posible evidenciar la rápida acumulación de cambios en su genoma. Esto supuso un cambio en el desarrollo y la mejora de los métodos diagnóstico, y fue de suma importancia en el desarrollo de vacunas y tratamientos. Desde la identificación inicial del SARS-CoV-2, hasta la fecha, se han compartido, a nivel mundial, más de 414.575 secuencias genómicas completas a través de bases de datos de acceso público. La capacidad de monitorizar la evolución viral casi en tiempo real tiene un impacto directo en la respuesta de salud pública a la pandemia de COVID-19 (17).
El SARS-CoV-2, al igual que el resto de los virus ARN, sufre cambios constantes en su genoma durante la replicación a través de mutaciones. La mayoría de las mutaciones no suponen una ventaja selectiva para el virus, ni tampoco cambios fenotípicos que impliquen alteraciones en el comportamiento o patrón de infección, sobre todo aquellas que puedan influir en la tasa de transmisión, el poder patógeno del virus o suponer un problema para el diagnóstico, las vacunas o los tratamientos actuales de la enfermedad. En cambio, algún cambio o varios si pueden suponer una ventaja, como un incremento en la transmisibilidad a través de un aumento en la unión al receptor (18–19).
Además, es preocupante la aparición de nuevas variantes que puedan asociarse con un aumento en la gravedad o en la letalidad, capaces de escapar al afecto de los anticuerpos neutralizantes que son generados tras una infección previa o tras la vacunación, y que se escapen a mediante los métodos diagnósticos actuales. La aparición de este tipo de variantes podría acarrear consecuencias epidemiológicas, generando un problema de salud pública, con repercusiones importantes en el control de la pandemia. Actualmente están consideradas variantes de preocupación (VOC): Alfa, Beta, Gamma y Delta (20,21,22,23). La variante dominante en estos momentos en España es la Delta, asociada a una mayor transmisibilidad y una ligera disminución de la efectividad vacunal. La variante Alfa ha descendido de forma considerable y las Beta y Gamma continúan detectándose con baja frecuencia. Excepto para el caso de Alfa (24), en el resto de variantes no se ha demostrado una mayor gravedad de los casos afectados. Por lo tanto, en la actualidad es importante poder identificarlas y hacer un seguimiento de las variantes circulantes en nuestro país.
Para poder tomar las medidas de salud pública que se consideren oportunas, es necesario la integración de los resultados de la secuenciación genómica en la vigilancia epidemiológica, para así poder detectar y monitorizar las variantes del SARS-CoV-2 (17).
Realizar un rastreo de la propagación del virus y conocer las posibles vías y dinámica de la transmisión, supone una ayuda en el seguimiento de la distribución geográfica y temporal de las mutaciones. Mediante el análisis filogenético, se puede conocer la historia evolutiva de un patógeno y por lo tanto proporcionar una gran información para orientar la respuesta frente a los brotes.
Descripción de la metodología para hacer secuenciación de genoma completo de SARS- CoV-2
La metodología de secuenciación del SARS- CoV-2 utilizada por los dos centros de referencia de Andalucía, se articula según los objetivos de vigilancia establecidos a nivel nacional (búsqueda de patrones de transmisión, asignación de clados / linajes, confirmación de reinfección, mutaciones fenotípicamente relevantes) lo que obliga al empleo de una secuencia de consenso del genoma completo o casi completo del virus. Aunque este abordaje, no presenta grandes problemas técnicos debido al tamaño del virus (aproximadamente 30.000 b), si está muy influenciado por la carga viral en la muestra (generalmente exudado nasofaríngeo) medida en base al valor de su umbral de ciclo (Cycle Threshold, CT), de forma que solo las muestras con valores CT menores a 30, son adecuadas para el objetivo establecido de secuenciación. El flujo de trabajo básico que se realiza se puede resumir de la siguiente manera:
- Preparación de muestras. Extracción del ARN y su conversión de ARN en ADNc (ADN complementario).
- Preparación de las librerías. Este proceso implica un flujo de trabajo de secuenciación basado en amplicones mediante el protocolo de código abierto ARTIC (https://artic.network/ncov- 2019).
- Secuenciación empleando plataformas por síntesis MiSeq/NextSeq (Illumina) o de semiconductores Ion-Torrent (Thermo-Fisher). Una vez concluida la secuenciación se obtiene un archivo de datos (FASTQ o BAM) para cada uno de los paired-ends, dicho archivo contiene las secuencias (reads) y los datos de calidad
- Análisis bioinformático. Este paso implica el procesamiento del archivo FASTQ o BAM para la obtención de la secuencia del genoma de consenso utilizando un pipeline (nf-core/ viralrecon (https://nf-co.re/viralrecon). La asignación de clados y las mutaciones anotadas se generan a través de la herramienta Nextclade (https://clades.nextstrain.org/). Posteriormente la herramienta de Pangolín (https://cov-lineages.org/) asigna linajes y para aquellos genomas donde no se puede obtener un linaje, se asigna un linaje imputado usando la herramienta interna impuSARS (25).
Automatización: ¿una necesidad no cubierta?
La NGS está en un proceso de evolución desde una tecnología utilizada con fines de investigación a una que se aplica en el diagnóstico clínico y epidemiológico. Esto implica, sobre todo en el caso epidemiológico, la necesidad de estudio de un alto número de muestras. En el escenario actual de la pandemia SARS-CoV-2, el número de muestras a secuenciar es muy alto, oscilando entre 400 y 600 semanales. Desde el punto de vista de la secuenciación propiamente dicha, esto no es un problema, ya que los secuenciadores de alto rendimiento utilizados ofrecen secuenciaciones totalmente automatizadas. Sin embargo, la compleja y engorrosa preparación de las librerías, es un cuello de botella significativo.
Sin embargo, los protocolos de preparación de librerías son procesos que son susceptibles de ser automatizados con desarrollos tecnológicos externos a las plataformas de secuenciación. La construcción de librerías de secuenciación es un proceso de varios pasos que podría ser robotizado, sobre todo: la tagmentación (fragmentación y etiquetado los amplicones), limpieza tras la tagmentación, la amplificación de los amplicones fragmentados, la agrupación y limpieza de las bibliotecas, por último la cuantificación y normalización de bibliotecas.
En base a lo anterior, los laboratorios de referencia han implementado una estrategia de automatización para la construcción de las librerías de secuenciación basadas en el empleo de plataformas robotizadas de código abierto OPENTRONS. Esta estrategia, de desarrollo propio, ha permitido aumentar la capacidad de secuenciación de forma escalable, logrando capacidades que pueden llegar a 1000 muestras/semana sin incrementar la dotación de personal. Además, este desarrollo ha permitido garantizar la normalización y la reproducibilidad en la preparación de las librerías de secuenciación, aspectos muy importantes cuando se trabaja con alto número de muestras.
Circuito asistencial e indicaciones de secuenciación en Andalucía
Desde la publicación por parte de la Dirección General de Salud Pública y Ordenación Farmacéutica de la Consejería de Salud y Familias de la “Instrucción para la secuenciación de SARS-CoV-2 en Andalucía”, se establecen dos centros de referencia para dar cobertura a la población de nuestra CCAA. Estos centros se ubican en el Servicio de Microbiología del Hospital Universitario Virgen del Rocío, en Sevilla, para dar cobertura a la población de Cádiz, Córdoba, Huelva y Sevilla, y en el Servicio de Microbiología del Hospital Universitario Clínico San Cecilio, para dar cobertura a la población de Almería, Granada, Jaén y Málaga.
La instrucción se alinea con las directrices marcadas por el Ministerio de Sanidad, Consumo y Bienestar social; en su ultima actualización, de fecha 06/07/2021, se reconocen las siguientes indicaciones:
1. Sospecha de reinfección. Definición de reinfección aportada por la Estrategia de detección precoz, vigilancia y control de COVID19.
2. Sospecha de nuevas variantes en casos confirmados con vínculos epidemiológicos con lugares o ámbitos de alta incidencia.
3. Casos con sospecha de infección con variantes que escapan a la inmunidad. Casos con sospecha de fallo vacunal. De igual manera, aunque no se considera fallo vacunal, se recomienda realizar secuenciación en aquellas personas asintomáticas que en el momento del diagnóstico han recibido una pauta de vacunación completa y ha transcurrido el tiempo estipulado según cada vacuna.
4. Situaciones en las que se sospeche una alta transmisibilidad o virulencia, como pueden ser brotes con un número de casos secundarios por caso muy alto o proporción de casos graves muy elevada. En este sentido, se actualiza el criterio para secuenciación de los casos graves que, además, incluirá a todo paciente hospitalizado en camas de UCI de los hospitales del SSPA.
Con respecto a este último punto, se priorizará la secuenciación en los casos índices de brotes epidémicos con una evolución fuera de lo esperado. Además, se priorizará la secuenciación del SARS CoV-2 en personas menores de 60 años sin antecedentes clínicos de comorbilidad asociados a mal pronóstico y cuya progresión clínica requiera de ingreso en Unidad de Cuidados Críticos, por ejemplo, por distrés respiratorio e insuficiencia respiratoria grave, en un corto período de evolución, menos de 3-4 días desde inicio de síntomas.
5. Todos los casos confirmados de COVID-19 con variantes de interés (VOC), distintas de B.1.1.7, a la prueba de PCR alélica específica
En consonancia con lo aprobado en el documento de Integración de la secuenciación genómica en la Vigilancia del SARS-CoV-2 del Ministerio de Sanidad se seleccionará una muestra aleatoria de las pruebas positivas recibidas semanalmente con Ct<30, correspondiendo aproximadamente un 80% de ellas a muestras procedentes de atención primaria (AP) y un 20% de atención hospitalaria (AH).
Andalucía cuenta con un circuito asistencial de secuenciación de SARS-CoV-2 único en nuestro país. Este circuito se basa en la colaboración y la coordinación multidisciplinar de diferentes áreas profesionales (asistencia sanitaria, microbiología, vigilancia en Salud Pública y bioinformática), con el apoyo del Sistema de Vigilancia Epidemiológica de Andalucía de la Dirección General de Salud Pública y Ordenación Farmacéutica y el Área de Bioinformática Clínica de la Fundación Progreso y Salud.
Las muestras que se reciben en los centros de referencia se procesan conforme a los protocolos descritos con anterioridad. El envío y selección de las muestras no aleatorias se realiza en coordinación con los servicios de dedicados a la vigilancia en Salud Pública (medicina preventiva y epidemiología).
Los archivos derivados de la secuenciación son analizados localmente y en el área de bioinformática de Fundación Progreso y Salud, que se encarga, además, del análisis filogenético de las secuencias.
En el área de bioinformática se reciben, en paralelo, los datos clínicos de los pacientes, a través de la base de datos poblacional de salud, lo que dota de un extraordinario valor a la información de la que se dispone.
Los hallazgos resultantes (cepa, variantes de interés -VOI- y de preocupación -VOC-) se integran con la información clínica y la información epidemiológica.
La figura 1 representa un esquema del circuito asistencial vigente en Andalucía. La descripción completa se puede consultar en http://www.clinbioinfosspa.es/COVID_circuit/.
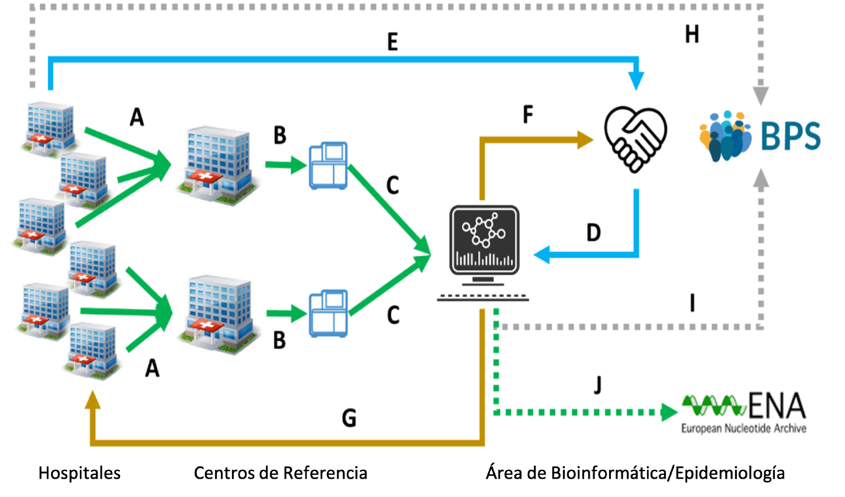
Figura 1.
Circulación de variantes de SARS-CoV-2 en Andalucía en 2021
Se presentan los datos de prevalencia y de evolución temporal desde la primera semana epidemiológica de 2021. En este periodo se han secuenciado 8573 muestras, de las cuales 5548 corresponden al centro de referencia del Hospital Universitario Virgen del Rocío, y 3025 al centro de referencia del Hospital Universitario Clínico San Cecilio.
La variante más prevalente durante este año 2021 ha sido la variante Alfa (B.1.1.7), de la que se detectaron 5994 casos, lo que supone el 70.3% del total. Le siguen por orden de prevalencia las variantes Delta (B.1.617.2) con un 18.8% de los casos (n=1606), la variante B.1.177 y sublinajes con un 4.6% de los casos (n=394), y la variante Gamma (P.1) con un 1.8% de los casos (n=151).
Se han detectado casos de un gran nº de variantes, que se presentan en la tabla 1.

Tabla 1. Distribución de las variantes analizadas hasta la fecha del artículo.
Respecto a la evolución temporal, la figura 2 representa la detección por semana epidemiológica de las variantes de preocupación con una mayor presencia en Andalucía.
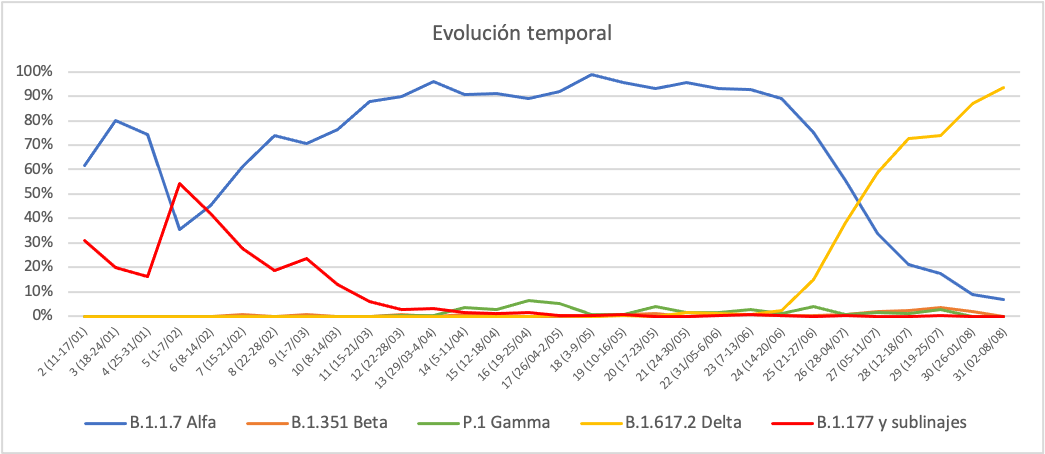
Figura 2.
Como se puede observar, la variante alfa comenzó su introducción en las primeras semanas de 2021 y, para la semana 12, desplazó a la cepa europea (linaje B.1.177). Es de destacar que en las semanas 2 a 5 los esfuerzos de secuenciación se dirigieron a la búsqueda de variante alfa, lo que explica la inversión de las curvas en estas semanas respecto a las posteriores. Las variantes beta y gamma no han logrado una introducción suficiente en Andalucía como para desplazar a la variante alfa, y no ha sido hasta la aparición de los primeros casos de variante delta (semana epidemiológica 24) cuando hemos asistido al inicio de un nuevo desplazamiento. En la actualidad la variante dominante en Andalucía es la variante delta.
La descripción detallada de los linajes aparecidos en el tiempo se puede consultar en http://nextstrain.clinbioinfosspa.es/SARS-COV-2-all?c=lineage; los principales resultados se muestran en la figura 3.

Figura 3.
CONCLUSIONES
La pandemia de COVID-19 ha acelerado la llegada de la secuenciación de genomas completos a los Servicios de Microbiología. En Andalucía se ha organizado un entorno asistencial único, que recoge los esfuerzos de los servicios de microbiología, los servicios de dedicados a la vigilancia en Salud Pública (medicina preventiva y epidemiología), de los centros de referencia para la secuenciación genómica de SARS-CoV-2, del área de bioinformática de la Fundación Progreso y Salud, del Servicio Andaluz de salud, y de la Dirección General de Salud Pública y Ordenación Farmacéutica, y que permite el control y evolución en tiempo real de las características virológicas de SARS-CoV-2. Esta iniciativa debe contemplarse para el abordaje de futuras pandemias y alertas de salud pública en enfermedades infecciosas.
REFERENCIAS BIBLIOGRÁFICAS
- ↑Goodwin S, McPherson JD, McCombie WR. Coming of age: ten years of next-generation sequencing technologies. Nat Rev Genet 2016;17:333–51. https://doi.org/10.1038/nrg.2016.49.
- ↑Carla López Causapé C, González Candelas F, Tomás Carmona M, Oliver Palomo A. Aplicaciones de las técnicas de secuenciación masiva en la Microbiología Clínica. 2021.71. Procedimientos en Microbiología Clínica. Sociedad Española de Enfermedades Infecciosas y Microbiología Clínica (SEIMC). 2021
- ↑Liu L, Li Y, Li S, Hu N, He Y, Pong R, et al. Comparison of Next-Generation Sequencing Systems. Journal of Biomedicine and Biotechnology 2012;2012:1–11. https://doi.org/10.1155/2012/251364.
- ↑Lu H, Giordano F, Ning Z. Oxford Nanopore MinION Sequencing and Genome Assembly. Genomics, Proteomics & Bioinformatics 2016;14:265–79. https://doi.org/10.1016/j.gpb.2016.05.004.
- ↑Quick J, Loman NJ, Duraffour S, Simpson JT, Severi E, Cowley L, et al. Real-time, portable genome sequencing for Ebola surveillance. Nature 2016;530:228–32. https://doi.org/10.1038/nature16996.
- ↑Next-Generation Sequencing for Infectious Disease Diagnosis and Management: A Report of the Association for Molecular Pathology. The Journal of Molecular Diagnostics 2015;17:623– 34. https://doi.org/10.1016/j.jmoldx.2015.07.004.
- ↑Lecuit M, Eloit M. The potential of whole genome NGS for infectious disease diagnosis. Expert Review of Molecular Diagnostics 2015;15:1517–9. https://doi.org/10.1586/14737159.2015.1111140.
- ↑Comas I, Cancino-Muñoz I, Mariner-Llicer C, Goig GA, Ruiz-Hueso P, Francés-Cuesta C, et al. Uso de las tecnologías de secuenciación masiva para el diagnóstico y epidemiología de enfermedades infecciosas. Enferm Infecc Microbiol Clin 2020;38:32–8. https://doi.org/10.1016/j.eimc.2020.02.006.
- ↑Gardy JL, Loman NJ. Towards a genomics-informed, real-time, global pathogen surveillance system. Nat Rev Genet 2018;19:9–20. https://doi.org/10.1038/nrg.2017.88.
- ↑Dessilly G, Goeminne L, Vandenbroucke A-T, Dufrasne FE, Martin A, Kabamba-Mukadi B. First evaluation of the Next-Generation Sequencing platform for the detection of HIV-1 drug resistance mutations in Belgium. PLoS One 2018;13:e0209561. https://doi.org/10.1371/journal.pone.0209561.
- ↑Fernández-Caso B, Fernández-Caballero JÁ, Chueca N, Rojo E, de Salazar A, García Buey L, et al. Infection with multiple hepatitis C virus genotypes detected using commercial tests should be confirmed using next generation sequencing. Sci Rep 2019;9:9264. https://doi.org/10.1038/s41598-019-42605-z.
- ↑Hendriksen RS, Bortolaia V, Tate H, Tyson GH, Aarestrup FM, McDermott PF. Using Genomics to Track Global Antimicrobial Resistance. Front Public Health 2019;0. https://doi.org/10.3389/fpubh.2019.00242.
- ↑Sabat AJ, Budimir A, Nashev D, Sá-Leão R, van Dijl JM, Laurent F, et al. Overview of molecular typing methods for outbreak detection and epidemiological surveillance. Eurosurveillance 2013;18. https://doi.org/10.2807/ese.18.04.20380-en.
- ↑World Health Organization. (2018). The use of next-generation sequencing technologies for the detection of mutations associated with drug resistance in Mycobacterium tuberculosis complex: technical guide. World Health Organization. https://apps.who.int/iris/handle/10665/274443.
- ↑Wagner MA, Wesmiller SW, Maydick M, Gawron LM, Peterson-Burch FM, Conley YP. Symptom Science: Omics and Response to Non-Pharmacological Interventions. Biological Research For Nursing 2021;23:394–401. https://doi.org/10.1177/1099800420975205.
- ↑Quick J, Loman NJ, Duraffour S, Simpson JT, Severi E, Cowley L, et al. Real-time, portable genome sequencing for Ebola surveillance. Nature 2016;530:228–32. https://doi.org/10.1038/nature16996.
- ↑Boehm E, Kronig I, Neher RA, Eckerle I, Vetter P, Kaiser L. Novel SARS-CoV-2 variants: the pandemics within the pandemic. Clinical Microbiology and Infection 2021;27:1109–17. https://doi.org/10.1016/j.cmi.2021.05.022.
- ↑CDC. Coronavirus Disease 2019 (COVID-19). Centers for Disease Control and Prevention 2020. https://www.cdc.gov/coronavirus/2019-ncov/variants/variant-info.html (acces- sed August 17, 2021).
- ↑Actualización Epidemiológica: Variantes de SARS- CoV-2 en las Américas – 24 de marzo de 2021 – OPS/ OMS | Organización Panamericana de la Salud. (n.d.). Retrieved August 15, 2021, from https://www.paho.org/es/documentos/actualizacion-epidemiologica-variantes-sars-cov-2-americas-24-marzo-2021
- ↑Organización Mundial de la Salud. (2021). Secuenciación del genoma del SARS-CoV-2 con fines de salud pública: orientaciones provisionales, 8 de enero de 2021. Organización Mundial de la Salud. https://apps.who.int/iris/handle/10665/338892. Licencia: CC BY-NC-SA 3.0 IGO
- ↑Genomic sequencing of SARS-CoV-2: a guide to implementation for maximum impact on public health. (n.d.). Retrieved August 15, 2021, from https://www.who.int/publications/i/item/9789240018440
- ↑Centro de Coordinación de Alertas y Emergencias Sanitarias. Variantes de SARS-CoV-2 de preocupación e interés para la salud pública en España. Evaluación Rápida de Riesgo, 5ª actualización, 6 de agosto de 2021
- ↑Funk T, Pharris A, Spiteri G, Bundle N, Melidou A, Carr M, et al. Characteristics of SARS-CoV-2 variants of con- cern B.1.1.7, B.1.351 or P.1: data from seven EU/EEA countries, weeks 38/2020 to 10/2021. Eurosurveillance 2021;26:2100348. https://doi.org/10.2807/1560-7917. ES.2021.26.16.2100348.
- ↑Davies NG, Jarvis CI, Edmunds WJ, Jewell NP, Diaz-Or- daz K, Keogh RH. Increased mortality in communi- ty-tested cases of SARS-CoV-2 lineage B.1.1.7. Nature 2021;593:270–4. https://doi.org/10.1038/s41586-021- 03426-1.
- ↑Ortuño FM, Loucera C, Casimiro-Soriguer CS, Lepe JA, Martinez PC, Diaz LM, et al. From partial to whole geno- me imputation of SARS-CoV-2 for epidemiological sur- veillance. BioRxiv 2021:2021.04.13.439668. https://doi.org/10.1101/2021.04.13.439668.
AGRADECIMIENTOS
A todos los centros hospitalarios que han contribuido con el envío de muestras para secuenciación.
Agradecemos especialmente al Secretario General de Investigación, Desarrollo e Innovación en Salud, el Dr. Isaac Túnez, su apoyo al establecimiento de los circuitos asistenciales de secuenciación en Andalucía.
INFORMACIÓN DEL ARTÍCULO
Conflicto de intereses: Los autores/as de este artículo declaran no tener ningún tipo de conflicto de intereses respecto a lo expuesto en el presente trabajo.
Autor para la correspondencia: Federico García. Servicio de Microbiología, Hospital Universitario Clínico San Cecilio, Granada Avenida Conocimiento, s/n · 18016, Granada, España. E-mail: fegarcia@ugr.es





