

Revisión
Actualización en el tratamiento de la neurodegeneración en las enfermedades lisosomales
Neurodegeneation treatment in lysosomal diseases. An update
Actual Med.2022;107(816):102-109 DOI: 10.15568/am.2022.816.rev01
Recibido: 03/06/2022
Revisado: 07/07/2022
Aceptado: 27/11/2022
RESUMEN
Las enfermedades lisosomales son un conjunto de enfermedades raras donde el déficit genético de determinadas enzimas da lugar a la acumulación de sustratos a nivel multiorgánico. El tratamiento de estos errores innatos del metabolismo sigue en continua investigación puesto que cuando existen, presentan limitaciones tratando de aumentar la síntesis de la enzima o disminuir el depósito de sustrato, sin llegar a la solución definitiva y, por tanto, a la curación. La neurodegeneración progresiva y limitante que la mayor parte de ellas producen desde edades pediátricas, supone uno de los mayores desafíos. El trasplante de células hematopoyéticas y la terapia de reemplazo enzimático son tratamientos aceptados para algunas de estas enfermedades; sin embargo, sus resultados sobre el control y la progresión de la sintomatología neurológicas en muchas ocasiones siguen siendo desalentadores. Sin duda, atravesar la barrera hematoencefálica es uno de los obstáculos que numerosos ensayos clínicos, que se están desarrollando en la actualidad, pretenden solventar. La terapia génica y aquella sobre las vías neurofisiológicas desencadenadas por la falta de degradación parcial o total de moléculas constituyen una nueva vía de estudio cuyo objetivo principal también se basa en evitar la progresión en el deterioro de la función cognitiva. Por tanto, el objetivo de este trabajo es realizar una revisión para conocer las terapias aprobadas y las líneas de investigación en curso en relación a la neurodegeneración de las enfermedades lisosomales. Es fundamental seguir avanzando en este campo con mayor desarrollo de estos enfoques y la búsqueda de nuevas perspectivas en las terapias que pueden suponer un gran impacto en la calidad de vida de estos pacientes.
Palabras clave: Enfermedades lisosomales; Neurodegeneración; Terapia de reemplazo enzimático; Trasplante de células hematopoyéticas; Terapia de genes..
ABSTRACT
Lysosomal diseases are a set of rare diseases where the genetic deficiency of certain enzymes gives rise to the accumulation of substrates at the multi-organ level. The treatment of these inborn errors of metabolism is still under continuous investigation since when they exist, they present limitations trying to increase the synthesis of the enzyme or decrease the deposit of substrate, without reaching the definitive solution, and therefore a cure. The progressive and limiting neurodegeneration that most of them produce from pediatric ages, is one of the greatest challenges. Hematopoietic cell transplantation and enzyme replacement therapy are accepted treatments for some of these diseases; however,its results on the control and progression of neurological symptoms often remain disappointing. Undoubtedly, crossing the blood-brain barrier is one of the obstacles that many clinical trials currently underway aim to overcome. Gene therapy and neurophysiological pathways triggered by the lack of partial or total degradation of molecules also constitute a new avenue of study whose main objective is also based on preventing progression in the deterioration of cognitive function. Therefore, the objective of this work is to carry out a review to know the approved therapies and the lines of research in progress in relation to the neurodegeneration of lysosomal diseases and It is essential to continue advancing in this field with further development of these approaches and the search new perspectives on therapies that can have a great impact on the quality of life of these patients.
Keywords: Lysosomal storage diseases; Neurodegeneration; Enzyme replacement therapy; Haematopoietic cell transplantation; Gene therapy.
Leer Artículo Completo
INTRODUCCIÓN
En las enfermedades por depósito lisosomal (EDL) existe una deficiencia de determinadas enzimas lisosomales que llevan a la acumulación de diferentes sustratos en múltiples órganos. En los últimos años, además del catabolismo, los lisosomas se han asociado a otras funciones que pretenden regular ciertos procesos celulares como el ciclo celular, el citoesqueleto, el transporte intracelular y la secreción de vesículas, el retículo endoplásmico, el aparato de Golgi, y vías de señalización, metabolismo, crecimiento, inmunidad, entre otros (1). Por ello, en la actualidad se hipotetiza que las manifestaciones clínicas de dichas enfermedades no solo son debidas al acúmulo de determinadas moléculas en tejidos y órganos, sino también al almacenamiento secundario de otros productos como gangliósidos, colesterol, ceramidas y esfingomielina, alteración de la autofagia, estrés oxidativo y anomalías de la homeostasis del calcio (1,2). El tratamiento dirigido a estas vías fisiopatológicas se encuentra en investigación. Algunas de las terapias estudiadas no han resultado eficaces, pero algunas otras están en desarrollo con importantes avances para evitar la progresión de estas enfermedades tan limitantes, la mayoría de ellas, ya desde edades tempranas de la vida.
Las EDL presentan afectación multiorgánica. En concreto, la disfunción lisosomal en la mucopolisacaridosis (MPS) altera la supervivencia de las neuronas debido a la acumulación de glucosaminoglucanos (GAG) (3). En la MPS II, se acumulan principalmente heparán sulfato (HS) y dermatán sulfato (DS). HS parece verse más implicada en la patología del sistema nervioso central (SNC) que DS debido a que las MPS que presentan discapacidad intelectual son aquellas en las que predomina su acumulación en lisosomas: tipos I, II,
III y VII (4). Se ha observado la participación de HS en la neurogénesis, la guía axonal y la sinaptogénesis por lo que su acumulación podría llevar a desajustes en estas vías. El principal desencadenante de la activación de la microglía es el HS mediante la unión a receptores tipo Toll. Incluso el que se acumula extracelularmente participa en este proceso al unirse a las moléculas de adhesión (2). En otras EDL como la lipofuscinosis ceroide neuronal tipo 2 (CLN2), el deterioro neurocognitivo se produce por el déficit de tripeptidil peptidasa 1 humana, y en la alfa-manosidosis, la deficiencia de la enzima alfa-manosidasa es responsable de la acumulación de manosilglicoproteínas en el SNC y otros órganos.
En la enfermedad de Niemann-Pick tipo C (NPC) los genes causales NPC1 (alrededor del 95% de los casos) y NPC2 (alrededor del 5% de los casos) conducen a la acumulación progresiva de colesterol no esterificado, esfingosina y glicoesfingolípidos. En otras como la enfermedad de Gaucher, la neuropatología, sobre todo periférica, se debe a la deficiencia en glucocerebrosidasa (GCasa).
El tratamiento de la neuropatología sigue siendo un reto en las EDL. Numerosos enfoques intentan mostrar la mejora de los síntomas neurológicos tales como trastorno cognitivo, hidrocefalia comunicante, compresión medular, hipertensión endocraneal, alteración de la deglución, hiperactividad, agresividad, alteración lenguaje (5). Actualmente, tanto la terapia de reemplazo enzimático (TRE) como el trasplante de células madre hematopoyéticas (TCMH) constituyen terapias aceptadas en algunas de ellas. Una de las limitaciones de la TRE es el traspaso de las enzimas a través de la barrera hematoencefálica (BHE). Por ello, se están estudiando numerosas vías de administración para limitar la neurodegeneración. Por otro lado, la
terapia génica se está desarrollando en diversos ensayos clínicos como opción definitiva para el control global de estas enfermedades.
Esta revisión se centra en describir la situación actual sobre el tratamiento dirigido a la estabilización y mejora de la función cognitiva de las EDL, así como de las investigaciones actualmente en desarrollo.
REVISIÓN. ACTUALIZACIÓN DE LAS TERAPIAS SOBRE LA NEURODEGENERACIÓN EN ENFERMEDADES LISOSOMALES
1. Terapia de reemplazo enzimático
Se basa en el uso de enzimas exógenas para que las células las internalicen a través de la vía del receptor de manosa. Es relativamente eficaz en el control de manifestaciones somáticas; sin embargo, las neurológicas con síntomas como déficit cognitivo, alteraciones del equilibrio y compresión de la médula espinal progresan en muchos casos. Esto se debe a la incapacidad de las enzimas recombinantes para cruzar la BHE. Actualmente, es el tratamiento de elección en algunas EDL, como en las MPS I, II, IV y VI (3).
Por ello, múltiples estudios están investigando diferentes vías de administración para asegurar el paso de las enzimas deficientes a través de la BHE, mostrando algunos de ellos resultados alentadores (1). Ciertas vías de administración, como intracerebroventricular (ICV) e intratecal (IT), se basan en introducir la enzima directamente al cerebro evitando así la BHE.
1.1. Administración intracerebroventricular
La α-N-acetil glucosaminidasa humana recombinante (rhNAGLU) es un enzima que ha mostrado disminuir la fosforilación de manosa 6. Debido a que el receptor manosa-6-fosfato (M6P) también es el receptor de IGF2 (factor de crecimiento similar a la insulina 2) en un sitio de unión diferente, la enzima rhNAGLU se fusiona con el receptor de IGF2 (rhNAGLU-IGF2) mejorando in vitro la orientación neuronal y astrocítica. Un ensayo clínico de fase I/II abierto tuvo como objetivo averiguar la dosis máxima de BMN-250 (rhNAGLUIGF2) en pacientes con MPS IIIB mediante infusión
ICV semanal. BMN-250 o tralesinidasa alfa normalizó las concentraciones de HS en el líquido cefalorraquídeo (LCR) en los 7 pacientes en 1 a 3 semanas de tratamiento con una adecuada tolerancia (2).
Para tratar el deterioro neurocognitivo de CLN2, se han realizado estudios con la tripeptidil peptidasa 1 humana recombinante o cerliponasa alfa, aprobada para esta enfermedad en 2017 por la Agencia Europea de Medicamentos (EMA) y la Administración de Medicamentos y Alimentos de Estados Unidos. Un estudio abierto la administró por vía intraventricular cada 2 semanas en pacientes de 3 y 16 años, mejorando el componente motor y la función del lenguaje, aunque como eventos adversos graves destacaron la infección de los dispositivos en 3 de los 23 pacientes, la movilización del dispositivo y las reacciones de hipersensibilidad (6).
1.2. Administración intratecal
La TRE IT a través de una inyección en la columna lumbar o en el espacio subaracnoideo en la cisterna magna mediante punción lumbar o un dispositivo de administración de fármacos IT (IDDD) se basa en el traspaso de pequeñas cantidades de enzima a través la capa ependimaria gracias a un gradiente de concentración (1,3). En ensayos clínicos de fase I/II, ha sido utilizada en pacientes con MPS I, II, IIIA y VI. En ellos, la corta vida media de la enzima, el riesgo de infecciones y respuestas inmunitarias hacia las enzimas y su poca eficacia han dificultado el tratamiento. Se
requirieron procedimientos invasivos y la implantación quirúrgica de dispositivos IT de administración de fármacos como el dispositivo SOPH-A-PORT mini S (1). No obstante, algún paciente con MPS I mostró mejorar sus síntomas de compresión medular y su función cognitiva en cuanto a memoria, atención y aprendizaje y los síntomas de la compresión medular mejoraron en un paciente con MPS VI (3).
En relación con los efectos adversos, en un ensayo de fase I/II abierto para pacientes con MPS IIIA donde se administró la enzima sulfamidasa humana recombinante (rhSGSH) a través de IDDD en 12 pacientes de 3 años, 7 pacientes experimentaron efectos adversos graves, 6 de ellos debido a movilización, desconexión o rotura de IDDD. Las concentraciones de HS y GAG en el LCR disminuyeron a pesar de formarse anticuerpos anti-rhSGSH en plasma en 6 de ellos. De ellos, 4 pacientes presentaron una disminución en las funciones cognitivas y 6 se estabilizaron. Un estudio posterior con
72 pacientes para establecer la dosis finalizó debido a que no se cumplían criterios de eficacia (2).
En 2014, comenzó un ensayo clínico de fase IIb, abierto, aleatorizado donde se administró rhSGSH a través de IDDD durante 48 semanas en niños con MPS IIIA en estadio temprano para evaluar la
seguridad y la eficacia. Se estableció que el fármaco era seguro y que todos los efectos adversos graves estaban relacionados con IDDD. No obstante, no se cumplieron los criterios de eficacia y el ensayo finalizó en 2016 (2).
El programa Therapeutics for Rare and Neglected Diseases de los Institutos Nacionales de la Salud en Estados Unidos (EEUU), está desarrollando la molécula 2-hidroxipropil-beta-ciclodextrina HPBeta-CD) para el tratamiento de las manifestaciones neurológicas de pacientes con NPC1. Actualmente, existe un estudio abierto de fase I/IIA de aumento de dosis de 2-hidroxipropil-β-ciclodextrinas (VTS-270) administrado por vía intratecal 36 meses en 14 pacientes de 4 a 23 años que padecían esta enfermedad, que ha mostrado una disminución del progreso de la enfermedad neurológica con respecto a la función cognitiva y el comportamiento adaptativo (7).
1.3. Administración intravenosa
La TRE IV es una vía en la que no está demostrado que la enzima atraviese la BHE en una cantidad adecuada para prevenir y tratar la clínica neurológica. Sin embargo, se están estudiando nuevas opciones como la denominada del “caballo de Troya” que se basa en unir la enzima recombinante con una molécula genéticamente modificada (proteínas de fusión quiméricas) que permita unirse a un receptor de la BHE (3).
Pabinafusp alfa (JR-141) está constituida por la fusión entre la enzima iduronato-2-sulfatasa humana y el extremo C-terminal de la pesada cadena del anticuerpo anti-receptor de transferrina humana. Es la primera y única enzima recombinante que puede administrarse con éxito por vía IV a través de la BHE. Tiene efectos beneficiosos tanto a nivel central como periférico. En un estudio, el desarrollo neurocognitivo fue casi normal en pacientes muy jóvenes que recibieron pabinafusp alfa, a diferencia de los pacientes mayores. El primer ensayo clínico de fase I/II en humanos en Japón con pacientes con MPS II mostró resultados prometedores. Posteriormente, el estudio de fase II en Brasil y el ensayo clínico de fase II/III en Japón confirmaron la eficacia a nivel central y periférico de pabinafusp alfa. Esto condujo a su aprobación en Japón en marzo de 2021 como la primera TRE en cruzar la BHE (8).
Actualmente se recomienda combinar la TRE con el TCMH ya que se ha demostrado que mejora el estado funcional del paciente antes del trasplante (3). En un estudio realizado en pacientes con MPS II tras haber sido tratados con idursulfasa (Elaprase®) por vía IV o TCMH, los niveles de HS y DS se normalizaron en la periferia, pero se encontraron elevados en LCR. La concentración de GAG y biomarcadores posteriores en el LCR y en el cerebro fue similar. La cadena ligera de neurofilamentos (Nf-L), un biomarcador utilizado para medir el daño axonal y la neurodegeneración a largo
plazo, también permaneció en altas concentraciones en el LCR y suero de estos pacientes a pesar de haber sido tratados con la TRE y el TCMH (9).
En enfermedades ultrararas como la alfa-manosidosis, la velmanase alfa (Lamzede®) es el único tratamiento autorizado en Europa para esta enfermedad lisosomal. Es una TRE vía IV que está aprobada para tratar síntomas no neurológicos en pacientes con enfermedad leve a moderada en Europa puesto que su eficacia a nivel neurológico no ha sido probada específicamente (10). Sin embargo, en el ensayo clínico con esta molécula en 19 niños y 14 adultos durante un máximo de 4 años, esta terapia mostró estabilizar y mejorar la función motora fina y gruesa en pacientes
pediátricos, más que en adultos, mediante la prueba integrada Bruininks-Oseretsky (BOT-2). Esta prueba mide la precisión de la motricidad fina, la integración de motricidad fina, la destreza manual, la coordinación bilateral, la coordinación de miembros superiores, la agilidad y la velocidad y, la fuerza. La subprueba “fuerza” no se evaluó; sin embargo, se han publicado resultados
positivos en los demás subtests (11).
2. Trasplante de células madre hematopoyéticas
Este procedimiento implica la entrega de células madre de donantes que producen la enzima implicada (3). Los monocitos y macrófagos donantes pueden cruzar la BHE e infiltrarse en el SNC donde pueden producir la enzima e influir en las manifestaciones neurológicas (12). Se considera actualmente el tratamiento de elección para los pacientes con MPS I menores de 2,5 años debido a los beneficios obtenidos en deterioro cognitivo, daño en el SNC, hidrocefalia y compresión de la médula espinal (3).
En la alfa-manosidosis, el TCMH está indicado para la prevención de muerte prematura y la desaceleración de la pérdida de la función cognitiva en edades muy precoces. En un estudio con una muestra muy limitada, 3 de 4 pacientes mejoraron sus funciones cognitivas, comportamiento adaptativo y memoria verbal. Incluso en el trasplante de células madre de sangre periférica sin células T, los resultados en un paciente fueron excelentes a nivel somático y neurológico (13). No hay estudios que comparen la eficacia entre el TCMH y la TRE; sin embargo, debido a que la EMA indicó que velmanase alfa no cruza la BHE, el TCMH podría seguir valorándose su papel en el tratamiento de la neurodegeneración (14).
3. Terapia génica
La terapia génica es una de las opciones que se está estudiando para tratar a estos pacientes con EDL. Consiste en la inyección del ADN de la enzima recombinante in vivo o ex vivo para que las células transducidas puedan sintetizar dichas enzimas. Para la administración de vectores virales, se han estudiado adenovirus, lentivirus y retrovirus (2,3). A diferencia de estos dos últimos, los adenovirus no tienden a integrarse en el genoma de la célula.
LYS-SAF302 es una terapia génica que usa el vector AAVrh10 para administrar el gen de la SGSH en el SNC en pacientes con MPS IIIA. Se encuentra en un ensayo de fase II/III establecido por primera vez en EEUU y se está expandiendo por Europa. Su objetivo es demostrar una mejora o normalización de la neuropatología después de 24 meses (15).
Otro ensayo clínico de fase I/II abierto está evaluando la seguridad y la eficacia de OTL-201, una terapia génica de células madre hematopoyéticas autólogas ex vivo, en 5 pacientes con MPS IIIA de 3 a 24 meses de edad. Antes de este estudio, un paciente se trató con busulfán seguido de un TCMH con células CD34+ autólogas transducidas con el vector LV.CD11b. hSGSH. Al año de este tratamiento, se demostró un injerto con niveles enzimáticos de leucocitos 25 veces superiores que el valor normal y una reducción de la concentración de HS en LCR, sangre y orina. Partiendo
de estos resultados, OTL-202 es un medicamento en investigación para MPS IIIB del que se esperan resultados prometedores (2).
En un estudio preliminar de fase I/II en Europa en MPS IIIB, 4 niños fueron tratados con la enzima NAGLU por vía intraparenquimatosa con mediante rAAV2/5 junto con fármacos inmunosupresores para evaluar su efecto en las manifestaciones neurológicas, así como para obtener información sobre la actividad enzimática en el LCR, la tolerancia y la inmunidad. La neuropatología mejoró notablemente, sobre todo, en el más joven (16).
4. Terapias con moléculas pequeñas
4.1. Terapia de reducción de sustrato
Esta opción terapéutica pretende inhibir la síntesis de sustrato para que así no se produzca su acumulación en los diferentes tejidos. Además, los inhibidores de la síntesis de sustratos son moléculas pequeñas que pueden cruzar la BHE. La vía del receptor del factor de crecimiento epidérmico (EGF) se encarga de regular la expresión de genes que codifican las enzimas responsables de la producción de GAG. La genisteína, que inhibe la activación de la tirosina quinasa del receptor EGF, fue la primera molécula utilizada para estudiar esta nueva vía de tratamiento. Sin embargo, en la actualidad no se han obtenido mejoras significativas en los síntomas neurológicos (1).
El miglustat, inhibidor de la enzima glucosilceramida sintasa, fue aprobado en 2009 por la EMA, y se utiliza vía oral para el tratamiento de los síntomas neurológicos progresivos en pacientes adultos y pediátricos con enfermedad de NPC, y en pacientes adultos con la enfermedad de Gaucher tipo 1 leve o moderada si no es adecuada la TRE (E: offlabel) (17). Actualmente, no se recomienda su uso en pacientes con enfermedad de Gaucher tipo 1 menores de 18 años porque no existe experiencia de su administración en ellos, aunque ya hay ensayos clínicos en proceso en pacientes pediátricos (18).
En NPC1, la resonancia magnética ha mostrado beneficio en el mantenimiento de la estructura anatómica subcortical y del cerebelo con miglustat. Estudios de cohortes con este fármaco han demostrado promover la estabilización de los síntomas neurológicos como trastornos de la marcha, lenguaje, deglución, entre otros, durante 2-8 años. Esta mejora es mayor en pacientes no pediátricos con inicio de las manifestaciones neurológicas (17).
4.2. Tratamiento a través de la lectura completa del codón parada
Numerosas pruebas moleculares demuestran que la mayoría de los sujetos con MPS I presentan una mutación prematura del codón parada en uno o ambos alelos del gen α-L-iduronidasa destacando
Q70X y W402X (19). Una de las vías terapéuticas para estos pacientes consiste en evitar el efecto del codón de parada prematuro promoviendo la síntesis de una proteína funcional. Estudios in vitro en células de pacientes con MPS indican que los aminoglucósidos, el cloranfenicol y el ataluren podrían utilizarse para este fin. Es esencial saber el genotipo de los pacientes puesto que esta estrategia depende de la mutación específica que causa la interrupción de la lectura del codón (3). La gentamicina es un aminoglucósido capaz de atravesar la BHE que se ha estudiado en pacientes con MPS I donde mejoró la lectura de la mutación Q70X (19). Ataluren se encuentra en un estudio de fase II en pacientes con MPS I cuyos objetivos son determinar el perfil de seguridad y calcular la farmacocinética de este fármaco en el LCR y en el plasma de estos pacientes (20).
4.3. Terapia con chaperonas
Se piensa que moléculas de pequeño tamaño, como las chaperonas, tienen más facilidades para cruzar la BHE (3). Este es el caso de ambroxol, una chaperona farmacéutica que recupera parte de la función de la enzima GCasa en la enfermedad de Gaucher (21).
4.4. Terapia con nanoliposoma
Actualmente, destaca un proyecto de investigación española en la red CIBERER para la enfermedad de Fabry. Es un producto liposomal de tercera generación (nanoliposoma) demostrando a nivel preclínico una eficacia mejorada frente a las TRE autorizadas del aporte de la enzima α-galactosidasa (22).
5. Terapia reguladora de la cascada inflamatoria
Los GAG alterados estructuralmente pueden activar el receptor tipo Toll (TLR) 4 debido a una reactividad cruzada frente al lipopolisacárido (LPS) de las bacterias gramnegativas. Esto conduce a la activación del factor de necrosis tumoral alfa (TNF-alfa) a través de su interacción con la proteína de respuesta primaria de diferenciación mieloide (MyD88) y la proteína de unión a LPS (LBP) (1,23). Esta vía de señalización también se puede activar mediante fragmentos de GAG derivados de la degradación de la matriz extracelular (23). Se han encontrado concentraciones elevadas de TNF-alfa en pacientes con MPS I, II y III. En el SNC, esto lleva a una inflamación cerebral crónica por lo que esta vía debe continuar en investigación pues podría constituir un enfoque terapéutico coadyuvante (1).
6. Regulación de la autofagia
Los lisosomas intervienen en la vía autofágica fusionándose con los autofagosomas y catabolizando las proteínas y los orgánulos contenidos en ellos mediante sus enzimas hidrolíticas (24). La alteración de este mecanismo se ha reconocido como una de las causas más relevantes en la neuropatología de algunas enfermedades lisosomales como en la MPS III y esto lleva al depósito de biomarcadores relacionados con la neurodegeneración tales como amiloide-β, tau y α-sinucleína, a la acumulación secundaria de materiales tóxicos como las proteínas poliubiquitinadas y a la
disfunción mitocondrial (2).
En estudios con células de MPS IIIA y deficiencia múltiple de sulfatasas, la sobreexpresión del gen que codifica el factor de transcripción EB, regulador de la autofagia, conlleva una eliminación casi total de los GAG (1).
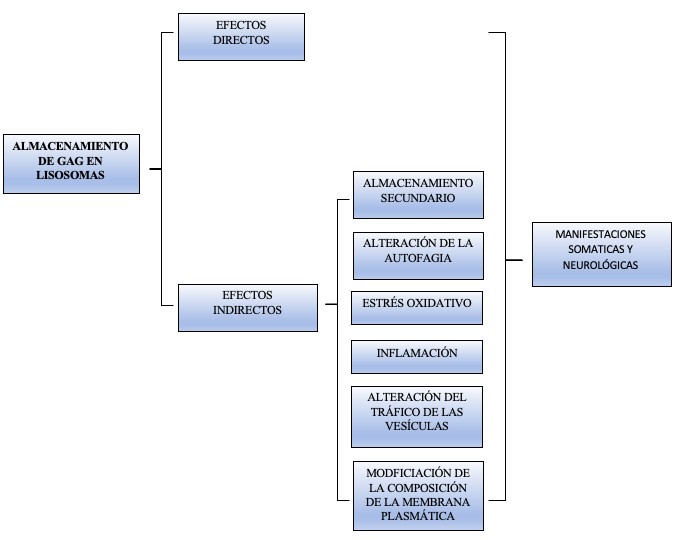
Figura 1. Consecuencias del almacenamiento de glucosaminoglucanos en mucopolisacaridosis. En las investigaciones de los últimos años, las manifestaciones clínicas dependen de la acumulación directa de glucosaminoglucanos y de la activación de vías secundarias.
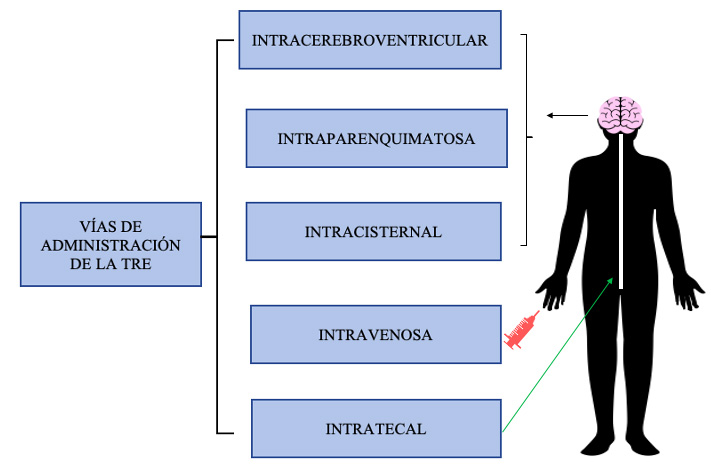
Figura 2. Vías de administración de la terapia de reemplazo enzimático para tratar la neurodegeneración en las enfermedades por depósito lisosomal. La terapia de reemplazo enzimático puede administrarse de forma directa al cerebro mediante las vías intratecal, intracerebroventricular, intraparenquimatosa e intracisternal. Las enzimas también pueden suministrarse vía intravenosa.
CONCLUSIONES
Actualmente, hay avances en el tratamiento de las EDLS, pero en muchos casos, sin efectos sobre la progresión neurológica. Este hecho tiene especial importancia puesto que la mayoría de las enfermedades lisosomales cursan con neurodegeneración ya desde edades tempranas. La calidad de vida de los pacientes y de sus familias se ve mermada por esta situación. La investigación es una pieza fundamental para seguir avanzando en este campo. El estudio de las distintas vías de administración de la TRE, de los vectores utilizados en la terapia génica, de nuevas vías neurofisiológicas, entre otros, son enfoques que pretenden tratar el deterioro cognitivo progresivo que sufren estos pacientes. A pesar de que muchos de ellos muestran resultados prometedores, son necesarios ensayos clínicos multicéntricos y estudios post-observacionales con mayor evidencia científica, utilizando protocolos consensuados internacionalmente.
REFERENCIAS BIBLIOGRÁFICAS
- ↑Fecarotta S, Gasperini S, Parenti G. New treatments for the mucopolysaccharidoses: from pathophysiology to therapy. Italian Journal of Pediatrics. 2018;44.
- ↑Yilmaz BS, Davison J, Jones SA, Baruteau J. Novel therapies for mucopolysaccharidosis typeIII. Journal of Inherited Metabolic Disease. 2021;44(1):129-47.
- ↑Scarpa M, Orchard PJ, Schulz A, Dickson PI, Haskins ME, Escolar ML, et al. Treatment of brain disease in the mucopolysaccharidoses. Molecular Genetics and Metabolism. 2017;122:25-34.
- ↑Morimoto H, Kida S, Yoden E, Kinoshita M, Tanaka N, Yamamoto R, et al. Clearance of heparan sulfate in the brain prevents neurodegeneration and neurocognitive impairment in MPS II mice. Molecular Therapy. 2021;29(5):1853-61.
- ↑Suarez-Guerrero JL, Higuera P, Florez JSA, Contreras-Garcia GA. Mucopolysaccharidosis: clinical features, diagnosis and management. Revista Chilena De Pediatria-Chile. 2016;87(4):295-304.
- ↑Schulz A, Ajayi T, Specchio N, de Los Reyes E, Gissen P, Ballon D, et al. Study of Intraventricular Cerliponase Alfa for CLN2 Disease. New England Journal of Medicine. 2018;378(20):1898-907.
- ↑Farmer CA, Thurm A, Farhat N, Bianconi S, Keener LA, Porter FD. Long-Term Neuropsychological Outcomes from an Open-Label Phase I/IIa Trial of 2-Hydroxypropyl-beta-Cyclodextrins (VTS-270) in Niemann-Pick Disease, Type C1. Cns Drugs. 2019;33(7):677-83.
- ↑Giugliani R, Martins AM, Okuyama T, Eto Y, Sakai N, Nakamura K, et al. Enzyme Replacement Therapy with Pabinafusp Alfa for Neuronopathic Mucopolysaccharidosis II: An Integrated Analysis of Preclinical and Clinical Data. International Journal of Molecular Sciences. 2021;22(20).
- ↑Bhalla A, Ravi R, Fang M, Arguello A, Davis SS, Chiu CL, et al. Characterization of Fluid Biomarkers Reveals Lysosome Dysfunction and Neurodegeneration in Neuronopathic MPS II Patients. International Journal of Molecular Sciences. 2020;21(15).
- ↑Hennermann JB, Guffon N, Cattaneo F, Ceravolo F, Borgwardt L, Lund AM, et al. The SPARKLE registry: protocol for an international prospective cohort study in patients with alpha-mannosidosis. Orphanet Journal of Rare Diseases. 2020;15(1).
- ↑Phillips D, Hennermann JB, Tylki-Szymanska A, Borgwardt L, Gil-Campos M, Guffon N, et al. Use of the Bruininks-Oseretsky test of motor proficiency (BOT-2) to assess efficacy of velmanase alfa as enzyme therapy for alpha-mannosidosis. Molecular Genetics and Metabolism Reports. 2020;23.
- ↑Gleitz HFE, Liao AY, Cook JR, Rowlston SF, Forte GMA, D’Souza Z, et al. Brain-targeted stem cell gene therapy corrects mucopolysaccharidosis type II via multiple mechanisms. Embo Molecular Medicine. 2018;10(7).
- ↑Ceccarini MR, Codini M, Conte C, Patria F, Cataldi S, Bertelli M, et al. Alpha-Mannosidosis: Therapeutic Strategies. International Journal of Molecular Sciences. 2018;19(5).
- ↑Naumchik BM, Gupta A, Flanagan-Steet H, Steet RA, Cathey SS, Orchard PJ, et al. The Role of Hematopoietic Cell Transplant in the Glycoprotein Diseases. Cells. 2020;9(6).
- ↑Registros de Ensayos Clínicos [Internet]. Clinicaltrialsregister.eu. [citado el 30 de abril de 2022]. Disponible en: https://www.clinicaltrials.gov/ct2/show/NCT03612869?term=NCT03612869&draw=2&rank=1
- ↑Tardieu M, Zerah M, Gougeon ML, Ausseil J, de Bournonville S, Husson B, et al. Intracerebral gene therapy in children with mucopolysaccharidosis type IIIB syndrome: an uncontrolled phase 1/2 clinical trial. Lancet Neurology. 2017;16(9):712-20.
- ↑Pineda M, Walterfang M, Patterson MC. Miglustat in Niemann-Pick disease type C patients: a review. Orphanet Journal of Rare Diseases. 2018;13.
- ↑Stirnemann J, Belmatoug N, Camou F, Serratrice C, Froissart R, Caillaud C, et al. A Review of Gaucher Disease Pathophysiology, Clinical Presentation and Treatments. International Journal of Molecular Sciences. 2017;18(2).
- ↑Kamei M, Kasperski K, Fuller M, Parkinson-Lawrence EJ, Karageorgos L, Belakhov V, et al. Aminoglycoside-Induced Premature Stop Codon Read-Through of Mucopolysaccharidosis Type I Patient Q70X and W402X Mutations in Cultured Cells. Jimd Reports, Vol 13. 2014;13:139-47.
- ↑Registros de Ensayos Clínicos [Internet]. Clinicaltrialsregister.eu. [citado el 30 de abril de 2022]. Disponible en: https://www.clinicaltrialsregister.eu/ctr-search/trial/2014-002596-28/DE#E
- ↑Ivanova MM, Dao J, Kasaci N, Adewale B, Nazari S, Noll L, et al. Cellular and biochemical response to chaperone versus substrate reduction therapies in neuropathic Gaucher disease. Plos One. 2021;16(10).
- ↑Tomsen-Melero J, Passemard S, Garcia-Aranda N, Diaz-Riascos ZV, Gonzalez-Rioja R, Pedersen JN, et al. Impact of Chemical Composition on the Nanostructure and Biological Activity of alpha-Galactosidase-Loaded Nanovesicles for Fabry Disease Treatment. Acs Applied Materials & Interfaces. 2021;13(7):7825-38.
- ↑Watts C. Lysosomes and lysosome-related organelles in immune responses. Febs Open Bio. 2022;12(4):678-93.
- ↑Lieberman AP, Puertollano R, Raben N, Slaugenhaupt S, Walkley SU, Ballabio A. Autophagy in lysosomal storage disorders. Autophagy. 2012;8(5):719-30.
INFORMACIÓN DEL LIBRO
Conflicto de intereses: Los autores/as de este artículo declaran no tener ningún tipo de conflicto de intereses respecto a lo expuesto en el presente trabajo.
Correspondencia: Mercedes Gil-Campos. Hospital Reina Sofía. Avda Menéndez Pidal sn · 14004 Córdoba. E-mail: mercedes_gil_campos@yahoo.es





